Rat Skeletal Muscle Microvascular Endothelial Cells
Cat.No.: CSC-C8729W
Species: Rat
Source: Skeletal Muscle
Cell Type: Endothelial Cell; Microvascular Cell
- Specification
- Background
- Scientific Data
- Q & A
- Customer Review
Rat skeletal muscle microvascular endothelial cells arise from the walls of microvascular vessels in experimental rats' skeletal muscle tissue. Since skeletal muscle is the most fundamental muscle that moves our bodies, it can be divided roughly into red muscle fibres and white muscle fibers. Rat skeletal muscle microvascular endothelial cells are an integral, important member of the microvascular wall of skeletal muscle and crucial to the proper function of the endothelial barrier, controlling the flow of blood and supporting the migration of immune cells. They are polygonal or spindle-shaped in their morphology, typical of endothelial cells. It has a thick layer of microvilli and invaginated vesicles covering the cell surface, and these structures are crucial to carrying fluids and encoding signals between cells.
On the physiological level, skeletal muscle microvascular endothelial cells are involved in angiogenesis, tissue repair and immune regulation. They might, for instance, release angiogenic chemicals like VEGF (vascular endothelial growth factor) to encourage the development of new blood vessels. Additionally, they are deeply involved in the regulation of immune cell migration, which is crucial to inflammatory reactions and tissue repair. Because of their physiological specificity in science, rat skeletal muscle microvascular endothelial cells have become extensively employed to study angiogenesis processes, drug screening and disease models – providing a powerful backbone for several scientific studies.
High Metabolic Substrate Load Induces Mitochondrial Dysfunction in Rat Skeletal Muscle Microvascular Endothelial Cells
Cardiovascular disease, majorly caused by lifestyle-related hyperglycemia and hyperlipidemia, involves endothelial dysfunction linked to mitochondrial defects and ROS production. Prior research predominantly focused on large vessel endothelial cells, but skeletal muscle-derived microvascular endothelial cells, characterized by adaptability to muscle activity, might differ metabolically.
Hansen et al. aimed to determine how acute and prolonged exposure to high metabolic substrate levels affects mitochondrial function in skeletal muscle-derived microvascular endothelial cells, hypothesizing that it alters mitochondrial function by reducing respiration and increasing ROS formation. Rat skeletal microvascular endothelial cells were incubated for 3 days with either 5 mM or 11 mM glucose, and mitochondrial measurements were done with regular glucose. Intact cells with 11 mM glucose showed 60% lower respiration and 4.6 times higher H2O2/O2 emission than those with 5 mM glucose (Fig. 1a and b). In permeabilized cells, respiration was 53% lower and H2O2/O2 emission was 2.8 times higher with 11 mM glucose (Fig. 1a and b). CS activity and lactate levels were unchanged between groups (Figs. 3b). When incubated with palmitic acid (PA), no difference in mitochondrial function was observed (Fig. 2). However, acute PA without glucose reduced coupling efficiency significantly. Elevated glucose after 3 days increased the mitochondrial leak-control ratio 3.1-fold, while elevated PA did so by 1.5-fold. In conclusion, these results showed that acute and prolonged lipid availability, as well as prolonged hyperglycemia, induces mitochondrial dysfunction as evidenced by lower mitochondrial respiration and enhanced H2O2/O2 emission. Elevated plasma substrate availability may lead to microvascular dysfunction in skeletal muscle by impairing endothelial mitochondrial function.
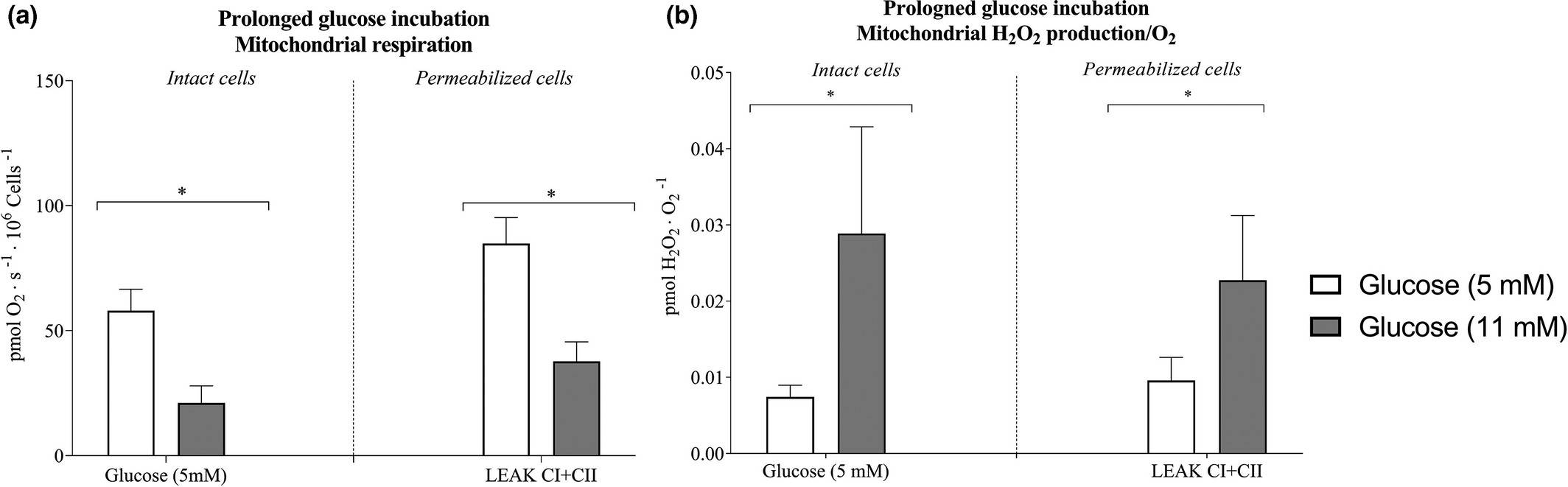 Fig. 1. Mitochondrial respiration and hydrogen peroxide (H2O2/O2) emission after 3-days prolonged glucose incubation of microvascular endothelial cells (Hansen C, Olsen K, et al., 2021).
Fig. 1. Mitochondrial respiration and hydrogen peroxide (H2O2/O2) emission after 3-days prolonged glucose incubation of microvascular endothelial cells (Hansen C, Olsen K, et al., 2021).
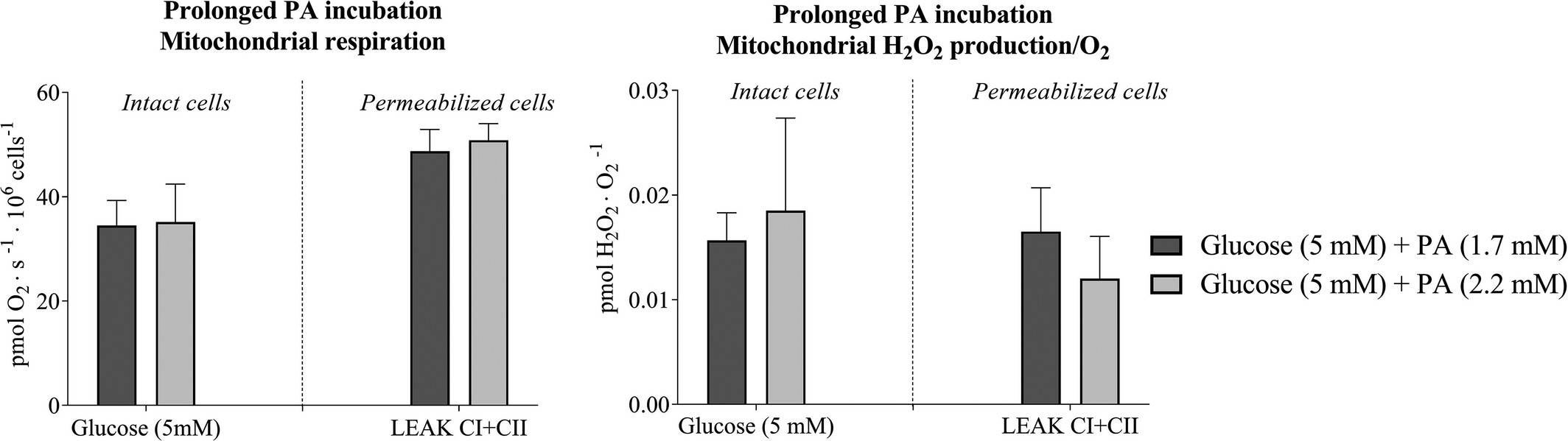 Fig. 2. Mitochondrial respiration and hydrogen peroxide (H2O2/O2) emission after 3-days prolonged palmitic acids incubation of microvascular endothelial cells (Hansen C, Olsen K, et al., 2021).
Fig. 2. Mitochondrial respiration and hydrogen peroxide (H2O2/O2) emission after 3-days prolonged palmitic acids incubation of microvascular endothelial cells (Hansen C, Olsen K, et al., 2021).
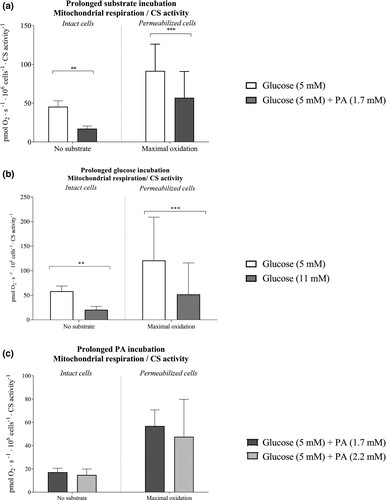 Fig. 3. Intrinsic mitochondrial respiration in microvascular endothelial cells after 3-day incubation with regular and elevated glucose or regular and elevated palmitic acid (PA) with 5 mM glucose concentrations (Hansen C, Olsen K, et al., 2021).
Fig. 3. Intrinsic mitochondrial respiration in microvascular endothelial cells after 3-day incubation with regular and elevated glucose or regular and elevated palmitic acid (PA) with 5 mM glucose concentrations (Hansen C, Olsen K, et al., 2021).
Vegfb Knockdown Attenuated CUG-Induced Muscle Lipid Accumulation and Insulin Resistance by Decreasing Lipid Uptake
Caloric restriction followed by refeeding, known as catch-up growth (CUG), leads to excessive lipid accumulation and insulin resistance in skeletal muscle, with the mechanisms poorly understood. CUG is correlated with obesity and type 2 diabetes, with lipid accumulation in muscle contributing to insulin resistance. Vascular endothelial growth factor B (VEGF-B) impacts lipid transport, potentially exacerbating fat accumulation and insulin resistance during CUG.
Lu's team used a rodent CUG model, examining VEGF-B signaling's impact on lipid transport through Vegfb knockdown in the tibialis anterior muscle. VEGF-B promotes lipid uptake and transport by upregulating FATP3 and FATP4 in some endothelial cells. In this study, rat primary skeletal muscle microvascular endothelial cells were isolated and cultured. Treatment with VEGF-B splice isoforms VEGF-B167 or VEGF-B186 increased Fatp3 and Fatp4 mRNA levels (Fig. 4A). Neutralizing antibodies against VEGFR1 or NRP1 reduced these levels (Fig. 4B), indicating VEGF-B's role through VEGFR1 and NRP1. Transfection with siRNA targeting Fatp3 or Fatp4 lowered FATP3 and FATP4 levels, reducing fatty acid uptake (Fig. 4C and D). VEGF-B186 treatment increased fatty acid uptake, which was counteracted by silencing Fatp3 or Fatp4 (Fig. 4E). VEGF-B186 enhanced fatty acid flux across endothelial layers without affecting permeability, which neutralizing antibodies blocked (Fig. 4F). VEGF-B signaling inhibition, therefore, reduces fatty acid uptake and transport in these cells.
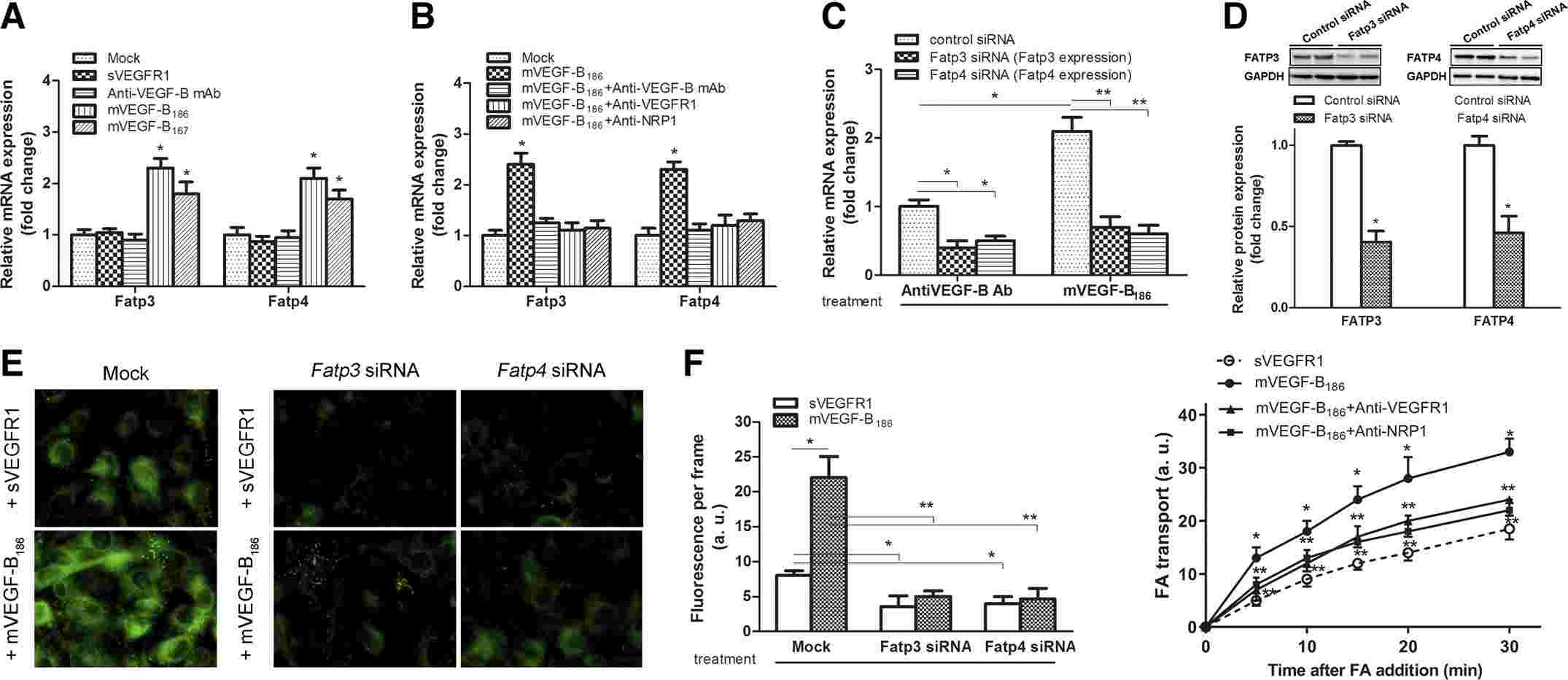 Fig. 4. Knockdown of Vegfb in the mice TA muscle attenuated CUG-induced muscle lipid accumulation and insulin resistance (Liu X, Hu S, et al., 2020).
Fig. 4. Knockdown of Vegfb in the mice TA muscle attenuated CUG-induced muscle lipid accumulation and insulin resistance (Liu X, Hu S, et al., 2020).
Ask a Question
Write your own review
Description: The thoracic aorta is located in the chest cavity and gives off arteries that branch to the esophagus, pericardium, lungs, and trachea. The thoracic aorta can be subdivided into the ascending aorta, ...
Description: Rat Podocytes are isolated from normal rat kidney. The cells are characterized by immunofluorescence with antibodies specific to podocin, Ang1, Nephrin, ACTN4, NPHS2. T25 flasks is required for cell ...
Description: Rat Bronchial Smooth Muscle Cells are isolated from normal rat bronchi tissue. Rat Bronchial Smooth Muscle Cells are characterized by immunofluorescence with antibodies specific to alpha-actin. T25 ...
Description: Guinea Pig Endothelial Cells from Creative Bioarray are isolated from guinea pig tissue. Prior to shipping, cells at passage 2 are detached from flasks and immediately cryopreserved in vials. Each ...
Description: Rat Lung Epithelial Cells are isolated from normal rat lung tissue. The cells are characterized by immunofluorescence with antibodies specific to CK-18, CK-19. T25 flasks is required for cell ...
Description: Rat Vein Endothelial Cells from Creative Bioarray are isolated from inferior vena cava tissue of 6-8 week old laboratory Sprague-Dawley rat. Rat Vein Endothelial Cells are grown in T75 tissue culture ...