Pancreatic Tumor Cells
- Background
- Applications
- Scientific Data
Pancreatic tumor cells arise from the abnormal growth and proliferation of cells within the pancreas. These cells can lose their normal regulatory mechanisms, leading to uncontrolled growth and the potential to invade surrounding tissues and metastasize to other parts of the body. The study of pancreatic tumor cells is a critical field within oncology and cancer biology, as understanding their behavior is essential for developing effective diagnostic tools, and therapeutic strategies, and, ultimately, improving patient outcomes.
Genetic and Epigenetic Alterations in Pancreatic Tumor Cells
- Key genetic mutations. At the molecular level, PDAC is heavily influenced by specific genetic mutations. The KRAS gene is mutated in over 90% of PDAC cases, often serving as an early event in tumorigenesis. Mutations in KRAS lead to uncontrolled cell proliferation and survival, highlighting its role as a critical oncogene. Notably, the RAS signaling pathway activated by these mutations promotes cancer cell growth and metastasis. Additionally, mutations in the TP53 gene are present in over 50% of pancreatic cancers. TP53 is crucial for maintaining genomic stability; its loss paves the way for further genetic alterations. Other mutations involving CDKN2A, which encodes a crucial tumor suppressor, and SMAD4, involved in the TGF-beta signaling pathway, are also commonly observed. These genetic alterations collectively disrupt the mechanisms regulating cell cycle progression and apoptosis, fostering a microenvironment conducive to cancer growth.
- Epigenetic factors. Beyond single-gene mutations, epigenetic changes play a pivotal role in pancreatic tumorigenesis. DNA methylation and histone modifications are key epigenetic mechanisms that can silence tumor suppressor genes without altering the underlying DNA sequence. For instance, hypermethylation of the CDKN2A promoter is frequently observed in PDAC, leading to the silencing of this critical cell cycle regulator. Furthermore, advancements in epigenetic profiling have revealed distinct patterns of global hypomethylation across various tumor types, including pancreatic cancer. These epigenetic alterations can foster an environment that supports tumor progression and metastasis by promoting pathways that evade normal cellular controls.
Research and Drug Development
The genetic and epigenetic landscape of pancreatic tumor cells serves as a foundation for innovative research and therapeutic strategies. Organoid cultures derived from patient tumors allow for the study of tumor biology in a more representative environment compared to traditional two-dimensional cell cultures. These organoids can be used to test drug efficacy, tailoring therapies to individual patients based on the unique genetic and epigenetic profiles of their tumors. Furthermore, advancements in CRISPR-Cas9 technology enable targeted gene editing of pancreatic tumor cells, facilitating the exploration of gene function in a precise manner.
Biomarker Discovery
The identification of biomarkers is crucial for early detection and monitoring of pancreatic cancer. Genetic alterations, particularly those in the KRAS gene, have been investigated as potential biomarkers for liquid biopsies. The detection of circulating tumor DNA (ctDNA) in patients can provide insights into the presence of disease and monitor treatment response. This non-invasive approach aligns with the growing need for early diagnostic tools, which are essential given the typically late-stage diagnosis of pancreatic cancer. Moreover, epigenetic changes, such as altered DNA methylation patterns, are being explored as biomarkers for early detection and prognostic assessment.
Mast Cells Induce Pancreatic Cancer Cell Proliferation and Invasion
After establishing that mast cells accumulate in pancreatic cancer in response to secreted cancer cell signals, mast cells' effect on pancreatic cancer cell proliferation and invasion was evaluated. Using conditioned media from LAD-2 cells, the proliferative effect of conditioned media was assessed compared to non-conditioned media on HPDE, PANC-1, and AsPC1 cells. There was no statistically significant change in HPDE proliferation with LAD-2 conditioned media vs. non-conditioned media at 24, 48, or 72 hours (Fig. 1a). In both PANC-1 and AsPC1 cells following 24, 48, and 72 hours of treatment with LAD-2-conditioned media there was a statistically significant increase in proliferation at all three-time points (p<0.01, p<0.001, p<0.001, and p<0.001, p<0.001, p<0.01 respectively for PANC-1 and AsPC1 cells at 24, 48, and 72 hours, Fig. 1b and c). In the AsPC1 proliferation assay, a decrease in thymidine incorporation at 72 hours compared to 48 hours may be attributed to contact inhibition.
Since the greatest effect on proliferation in both cancer cell lines was 48 hours, this time point was chosen to perform Boyden chamber Matrigel invasion assays. LAD-2 conditioned media induced a statistically significant increase in Matrigel invasion in both PANC-1 (13.0 +/- 1.0 vs. 7.3 +/- 0.6 cells per 200× high power field, p<0.05) and AsPC1 (88.3 +/- 11.0 vs. 19.7 +/- 0.6 cells per 200× high power field, p<0.01) cancer cells (Fig. 2). Of note, the HPDE cells demonstrated no Matrigel invasion at 48 hours with both non-conditioned and LAD-2 conditioned media.
To evaluate whether the effect of mast cell conditioned media on pancreatic cancer cell invasion was matrix metalloproteinase (MMP) dependent, AsPC1 cells were treated with either control media, mast cell conditioned media, or mast cell conditioned media with a broad spectrum MMP inhibitor, GM6001. The addition of GM6001 to the mast cell conditioned media led to a statistically significant reduction in invasion compared to conditioned media treatment. [(51.6+/-0.8 vs. 174.3+/-22.2 cells per 200× high power field, p<0.01, (Fig. 2g)]. Zymography of the mast cell conditioned media did not demonstrate the presence of MMP-2 or MMP-9.
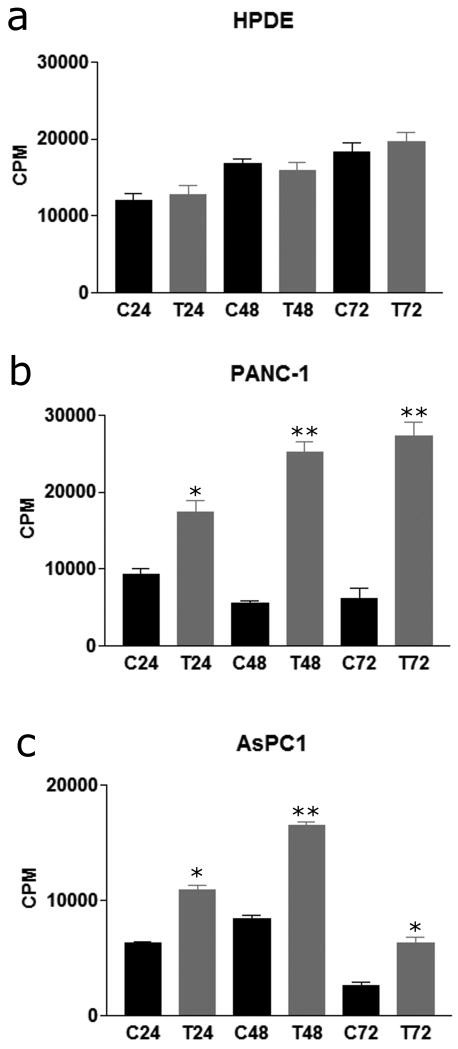 Fig. 1 Mast cells induce pancreatic
cancer cell proliferation. (Strouch MJ, et al, 2010)
Fig. 1 Mast cells induce pancreatic
cancer cell proliferation. (Strouch MJ, et al, 2010)
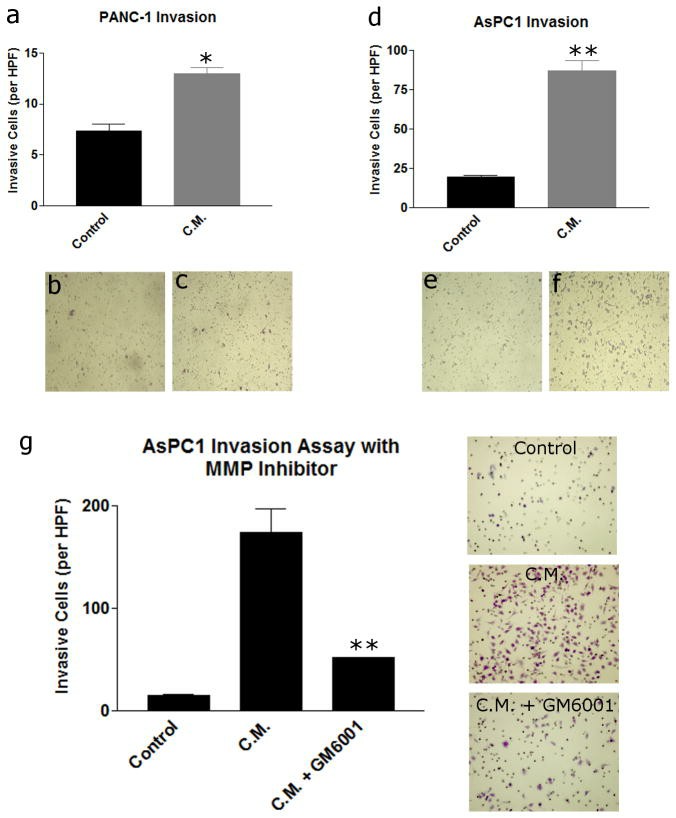 Fig. 2 Mast cell
conditioned media induces pancreatic cancer invasion through an MMP-dependent mechanism. (Strouch MJ,
et al, 2010)
Fig. 2 Mast cell
conditioned media induces pancreatic cancer invasion through an MMP-dependent mechanism. (Strouch MJ,
et al, 2010)
ROCK1 Knockdown Inhibits Pancreatic Cancer Cell Growth
ROCK, or Rho-associated coiled coil-containing protein kinase, is a member of the AGC kinase family and has been shown to play a role in cell migration, ECM synthesis, stress-fiber assembly, and cell contraction. The expression of ROCK1 was compared among multiple, established pancreatic cancer cell lines, as well as the immortalized normal pancreatic ductal epithelial cell line HPDE6 and the CAF cells (CW-1) isolated from a PDAC patient's tumor. Western blotting analysis showed robust expression among the PANC-1, Mia PaCa-2, and SU.86.86 cell lines, and relatively low expression among the BxPC3, AsPC-1, and HS766T lines (Fig. 3A). The HPDE6 cells also expressed a low level of ROCK1. The CW-1 CAFs did not express significant levels of full-length ROCK1 but rather appeared to express a ROCK1 antibody reactive protein of approximately 130 kDa, consistent with the molecular weight of a caspase-3 cleavage-activated ROCK1 fragment. To study the direct effects of ROCK1 expression, siRNA oligonucleotides were used to knock down its expression in the PANC-1 and SU.86.86 cell lines (Fig. 3B and 3C). ROCK1 expression continued to decline over 72 hours following siRNA treatment (Fig. 3B). ROCK1 siRNA treatment reduced the proliferation rate of PDAC cells. With time-course cell viability assays, decreased cell proliferation coincided with ROCK1 levels, where significant decreases in the proliferation of PANC-1 and SU.86.86 cells were not observed until, or after, approximately 72 hours (Fig. 3D). This effect was observed with multiple siRNAs and in two different cell lines.
To compare siRNA knockdown results with enzymatic inhibition using small molecules, the effects of the ROCK inhibitor, fasudil, were tested on the cell proliferation of several cell lines using the SRB assay (Fig. 4A). Fasudil is a well-described ROCK inhibitor that has shown clinical activity in treating cerebral vasospasm. Consistent with our siRNA data, fasudil showed a modest effect on pancreatic cancer cell growth, and minimal effect on CAF cell growth, with IC50s in the 50–80 μM range. ROCK1 siRNA also significantly inhibited PANC-1 tumor cell migration (Fig. 4B). To evaluate the effects of ROCK1 inhibition in a model more relevant to the in vivo environment, in which both tumor cells and CAFs are grown together, co-cultured SU.86.86 cells with CW-1 cells and the effects of fasudil on phenotypic properties of the cultured cells were tested. Using immunofluorescence microscopy (Fig. 4C), α-SMA and Collagen I expression in co-cultured cells treated with a range of fasudil concentrations for 72 hours. DNA staining reveals that significant numbers of α-SMA-negative cells (tumor cells) remain on the slide with even high concentrations of fasudil. α-SMA-positivity (CAFs) was largely lost following fasudil treatment in the context of co-culture, despite showing similar IC50 values in monoculture with fasudil treatment. Monocultured CAFs treated with fasudil also show some loss of α-SMA positivity, suggesting some reversion of CAF activation may be occurring (Fig. 4D). Importantly, this reduction in CAF cell proliferation or activation coincided with a reduction in total Collagen I expression, whether assessed qualitatively by microscopy, or semi-quantitatively by dot-blotting under non-denaturing conditions (Fig. 4E).
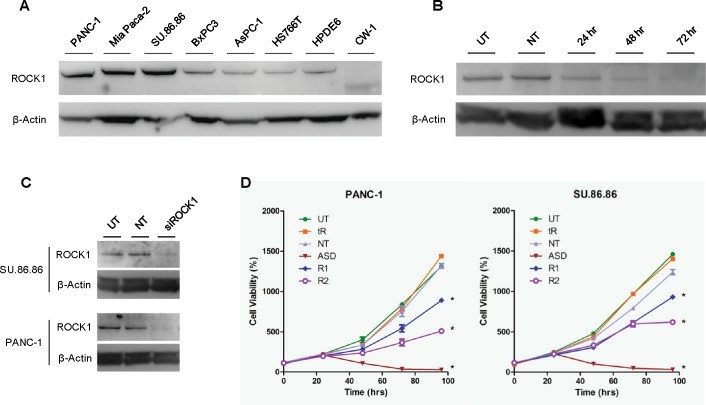 Fig.
3 Knockdown of ROCK1 by siRNA in pancreatic cancer cells inhibits cell proliferation. (Whatcott CJ,
et al., 2017)
Fig.
3 Knockdown of ROCK1 by siRNA in pancreatic cancer cells inhibits cell proliferation. (Whatcott CJ,
et al., 2017)
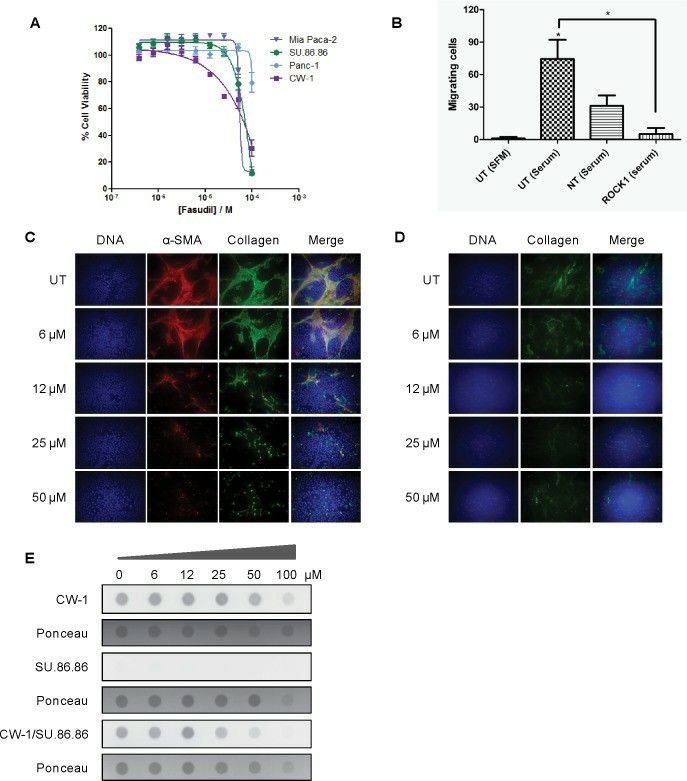 Fig.
4 Effects of the ROCK1 inhibition on pancreatic cancer cells and cancer-associated fibroblasts. (Whatcott CJ,
et al., 2017)
Fig.
4 Effects of the ROCK1 inhibition on pancreatic cancer cells and cancer-associated fibroblasts. (Whatcott CJ,
et al., 2017)
Filters Clear all filters
Species
- Cat (1)
- Human (992)
- Mouse (5)
- Rat (1)
Source
- Abdomen Metastasis (2)
- Adrenal Gland (7)
- Adrenal Gland Metastasis (2)
- Ascites (23)
- Ascites Metastasis (32)
- Bile Duct (3)
- Bladder (12)
- Blood (120)
- Bone (21)
- Bone Marrow (43)
- Bone Marrow Metastasis (18)
- Bone Metastasis (6)
- Brain (31)
- Brain Metastasis (6)
- Breast (8)
- Bronchus (1)
- Cecum (3)
- Cerebrospinal Fluid (1)
- Cerebrospinal Fluid Metastasis (1)
- Cervix (32)
- Colon (84)
- Cornea (3)
- Cutaneous Metastasis (1)
- Dermis (1)
- Duodenum (1)
- Endometrium (17)
- Esophagus (44)
- Eye (12)
- Eye Socket (5)
- Fetus (1)
- Foreskin (4)
- Gallbladder (1)
- Gingiva (2)
- Globe (2)
- Groin (1)
- Hypodermis Metastasis (5)
- Intestine (85)
- kidney (1)
- Kidney (9)
- Liver (13)
- Liver Metastasis (17)
- Lung (42)
- Lung Metastasis (8)
- Lymph Node (5)
- Lymph Node Metastasis (58)
- Muscle (4)
- Muscle Metastasis (2)
- Nose (2)
- Omentum Metastasis (2)
- Oral Cavity (10)
- Ovary (13)
- Ovary Metastasis (2)
- Pancreas (10)
- Pelvic Wall Metastasis (1)
- Pelvis (1)
- Perianal Space Metastasis (1)
- Pericardial Effusion (1)
- Pericardial Effusion Metastasis (1)
- Perineus (1)
- Peripheral Blood (119)
- Peritoneal Effusion (2)
- Peritoneum (1)
- Peritoneum Metastasis (1)
- Pharynx (3)
- Pleural Effusion (54)
- Pleural Effusion Metastasis (44)
- Prostate (4)
- Rectum (13)
- Renal Pelvis (1)
- Retroperitoneal Space (2)
- Salivary Gland (2)
- Skeletal Muscle (1)
- Skin (22)
- Skin Metastasis (3)
- Small Intestine (1)
- Small Intestine Metastasis (1)
- Soft Tissue Metastasis (1)
- Stomach (4)
- Testis (9)
- Thoracic Cavity Metastasis (6)
- Thyroid Gland (15)
- Thyroid Gland Metastasis (1)
- Tongue (5)
- Umbilical Cord (1)
- Umbilical Cord Blood (1)
- Urachus (1)
- Ureter (1)
- Uterus (53)
- Uvea (2)
- Vagina (2)
- Vulva (1)
Disease
- Acute Biphenotypic Leukemia (1)
- Acute Erythroid Leukemia (4)
- Acute Megakaryoblastic Leukemia (4)
- Acute Monocytic Leukemia (9)
- Acute Myeloid Leukemia (25)
- Acute Promyelocytic Leukemia (2)
- Adrenal Gland Neuroblastoma (11)
- Adult B Acute Lymphoblastic leukemia (1)
- Adult B Acute Lymphoblastic Leukemia (6)
- Adult T Acute Lymphoblastic Leukemia (6)
- Adult T Lymphoblastic Lymphoma (2)
- Adult T-Cell Leukemia/Lymphoma (1)
- Alveolar Rhabdomyosarcoma (4)
- Alveolar Ridge Squamous Cell Carcinoma (1)
- Amelanotic Melanoma (3)
- Ampulla of Vater Adenocarcinoma (1)
- Ampulla of Vater Adenosquamous Carcinoma (3)
- Anaplastic Astrocytoma (3)
- Anaplastic Large Cell Lymphoma (7)
- Askin Tumor (1)
- Astrocytoma (5)
- B Acute Lymphoblastic Leukemia (2)
- B-Cell Non-Hodgkin Lymphoma (5)
- Bare Lymphocyte Syndrome Type 2 (1)
- Barrett Adenocarcinoma (2)
- Benign Prostatic Hyperplasia (1)
- Bladder Carcinoma (12)
- Bladder Squamous Cell Carcinoma (1)
- Breast Adenocarcinoma (1)
- Breast Carcinoma (9)
- Breast Ductal Carcinoma (2)
- Burkitt Lymphoma (17)
- Canavan Disease (1)
- Cecum Adenocarcinoma (3)
- Central Nervous System Lymphoma (2)
- Cervical Adenocarcinoma (2)
- Cervical Adenosquamous Carcinoma (2)
- Cervical Small Cell Carcinoma (1)
- Cervical Squamous Cell Carcinoma (2)
- Childhood B Acute Lymphoblastic Leukemia (13)
- Childhood T Acute Lymphoblastic Leukemia (16)
- Childhood T Lymphoblastic Lymphoma (1)
- Cholangiocarcinoma (2)
- Chronic Eosinophilic Leukemia (1)
- Chronic Lymphocytic Leukemia (2)
- Chronic Myeloid Leukemia (23)
- Clear Cell Renal Cell Carcinoma (2)
- Colon Adenocarcinoma (55)
- Colon Carcinoma (34)
- Colorectal Adenocarcinoma (1)
- Colorectal Carcinoma (1)
- Congenital Pure Red Cell Aplasia (1)
- Cutaneous Melanoma (10)
- Dedifferentiated Chondrosarcoma (1)
- Desmoplastic Melanoma (1)
- Diffuse Large B-Cell Lymphoma (28)
- Down Syndrome (2)
- EBV-Related Burkitt Lymphoma (12)
- Embryonal Carcinoma (3)
- Embryonal Rhabdomyosarcoma (3)
- Endometrial Adenocarcinoma (13)
- Endometrial Adenosquamous Carcinoma (2)
- Endometrial Carcinoma (2)
- Endometrioid Stromal Sarcoma (1)
- Epithelioid Hemangioendothelioma (1)
- Epithelioid Sarcoma (3)
- Esophageal Adenocarcinoma (6)
- Esophageal Squamous Cell Carcinoma (41)
- Essential Thrombocythemia (1)
- Ewing Sarcoma (2)
- Extraskeletal Myxoid Chondrosarcoma (1)
- Fanconi Anemia (1)
- Fibrosarcoma (1)
- Follicular Lymphoma (2)
- Gallbladder Carcinoma (2)
- Gallbladder Undifferentiated Carcinoma (2)
- Gastric Adenocarcinoma (6)
- Gastric Adenosquamous Carcinoma (1)
- Gastric Carcinoma (5)
- Gastric Choriocarcinoma (1)
- Gastric Fundus Carcinoma (1)
- Gastric Signet Ring Cell Adenocarcinoma (1)
- Gastric Small Cell Carcinoma (2)
- Gastric Tubular Adenocarcinoma (5)
- Gastroesophageal Junction Adenocarcinoma (1)
- Gestational Choriocarcinoma (1)
- Gingival Squamous Cell Carcinoma (2)
- Glioblastoma (18)
- Gliosarcoma (1)
- Hairy Cell Leukemia (1)
- Hepatoblastoma (2)
- Hepatocellular Carcinoma (6)
- Hepatosplenic T-Cell Lymphoma (2)
- Hereditary Thyroid Gland Medullary Carcinoma (1)
- High Grade B-Cell Lymphoma (1)
- High Grade Ovarian Serous Adenocarcinoma (8)
- Hodgkin Lymphoma (9)
- Hypopharyngeal Squamous Cell Carcinoma (2)
- Infectious Mononucleosis (1)
- Intrahepatic Cholangiocarcinoma (6)
- Invasive Breast Carcinoma of No Special Type (12)
- Kidney Neoplasm (1)
- Kidney Rhabdoid Tumor (1)
- Krukenberg Tumor (1)
- Liposarcoma (1)
- Lung Adenocarcinoma (17)
- Lung Giant Cell Carcinoma (8)
- Lung Large Cell Carcinoma (9)
- Lung Mucoepidermoid Carcinoma (1)
- Lung Non-Small Cell Carcinoma (2)
- Lung Small Cell Carcinoma (25)
- Lung Squamous Cell Carcinoma (9)
- Lymphoblastic Lymphoma (1)
- Malignant Peripheral Nerve Sheath Tumor (1)
- Mantle Cell Lymphoma (5)
- Mature Gastric Teratoma (1)
- Maxillary Sinus Squamous Cell Carcinoma (1)
- Medulloblastoma (3)
- Melanoma (24)
- Meningioma (2)
- Minimally Invasive Lung Adenocarcinoma (1)
- Monophasic Synovial Sarcoma (1)
- Mouse Intestinal Tract Neuroendocrine Adenoma (1)
- Mouse Mammary Gland Malignant Neoplasm (2)
- Mouse Plasmacytoma (1)
- Mycosis Fungoides (1)
- Myelodysplastic Syndrome (1)
- Myxofibrosarcoma (1)
- Natural Killer Cell Lymphoblastic Leukemia/Lymphoma (2)
- Neuroblastoma (26)
- Oral Cavity Squamous Cell Carcinoma (15)
- Osteoid Osteoma (1)
- Osteosarcoma (15)
- Ovarian Carcinoma (1)
- Ovarian Clear Cell Adenocarcinoma (1)
- Ovarian Endometrioid Adenocarcinoma (4)
- Ovarian Granulosa Cell Tumor (1)
- Ovarian Mucinous Adenocarcinoma (2)
- Ovarian Serous Adenocarcinoma (2)
- Ovarian Serous Cystadenocarcinoma (2)
- Ovarian Small Cell Carcinoma (1)
- Pancreatic Adenocarcinoma (13)
- Pancreatic Carcinoma (5)
- Pancreatic Ductal Adenocarcinoma (12)
- Papillomavirus-Independent Cervical Squamous Cell Carcinoma (1)
- Papillomavirus-Related Cervical Adenocarcinoma (7)
- Papillomavirus-Related Cervical Squamous Cell Carcinoma (4)
- Papillomavirus-Related Endocervical Adenocarcinoma (16)
- Paroxysmal Nocturnal Hemoglobinuria (3)
- Pharyngeal Squamous Cell Carcinoma (1)
- Plasma Cell Myeloma (15)
- Pleural Epithelioid Mesothelioma (5)
- Pleural Sarcomatoid Mesothelioma (2)
- Poorly Differentiated Thyroid Gland Carcinoma (1)
- Primary Cutaneous T-Cell Non-Hodgkin Lymphoma (1)
- Primary Effusion Lymphoma (7)
- Primitive Neuroectodermal Tumor (1)
- Prostate carcinoma (1)
- Prostate Carcinoma (9)
- Rectal Adenocarcinoma (13)
- Rectosigmoid Adenocarcinoma (1)
- Recurrent Bladder Carcinoma (1)
- Renal Cell Carcinoma (7)
- Renal Pelvis Urothelial Carcinoma (1)
- Retinoblastoma (11)
- Sacral Chordoma (1)
- Sacrococcygeal Teratoma (1)
- Salivary Gland Squamous Cell Carcinoma (1)
- Sezary Syndrome (1)
- Shwachman-Diamond Syndrome (1)
- Skin Squamous Cell Carcinoma (2)
- Splenic Marginal Zone Lymphoma (1)
- Testicular Embryonal Carcinoma (8)
- Testicular Teratoma (2)
- Testicular Yolk Sac Tumor (1)
- Thyroid Gland Anaplastic Carcinoma (10)
- Thyroid Gland Follicular Carcinoma (4)
- Thyroid Gland Papillary Carcinoma (3)
- Thyroid Gland Sarcoma (1)
- Thyroid Gland Squamous Cell Carcinoma (2)
- Tongue Adenosquamous Carcinoma (1)
- Tongue Squamous Cell Carcinoma (6)
- Type I Endometrial Adenocarcinoma (1)
- Ureter Urothelial Carcinoma (1)
- Uterine Carcinosarcoma (2)
- Uterine Corpus Leiomyosarcoma (1)
- Uterine Corpus Sarcoma (2)
- Uveal Melanoma (2)
- Vaginal Melanoma (2)
- Vulvar Melanoma (1)
- Vulvar Squamous Cell Carcinoma (1)
Description: Established in 1985 from the liver metastasis of a primary pancreatic adenocarcinoma from a ...
Description: Established in 1985 from the primary ductal pancreatic adenocarcinoma (grade II) from a 44-year-old ...
Description: Established in 1985 from the liver metastasis of a primary pancreatic adenocarcinoma from a ...
Description: Established from the malignant ascites of a 60-year-old Japanese man with pancreas carcinoma in 1985
Description: Established from the malignant ascites of a 66-year-old Japanese man with pancreas carcinoma in 1984
Description: Established from ascites obtained during exploratory laparatomy of a 43-year-old Japanese man with ...
Description: Originally described to be derived from the moderately differentiated adenocarcinoma of the head of ...
Description: Species: human; Tissue: pancreas; Tumor: adenocarcinoma
Description: established from the ascites metastasis from a 47-year-old female with pancreatic ductal ...
Description: Established in 2012 from a primary pancreatic tumor of a 78-year-old Caucasian man with ductal ...
Description: Established in 2012 from a liver metastasis of a 63-year-old Caucasian woman with ductal ...
Description: Established in 2010 from the pancreatic tumor of a 46-year old man with poorly differentiated ...
Description: Pancreatic adenocarcinoma producing CEA. K-ras activated. Cell growth is slow.
Description: Pancreatic ductal cell carcinoma. PTHrP producing.
Description: Pancreatic ductal cell carcinoma. PTHrP producing.
Description: Pancreatic ductal cell carcinoma. PTHrP producing.
Description: Human pancreatic cancer cell line establesh from liver metastasis.
Description: Human tumor cell line secreting parathyroid hormone-related peptide (PTHrP) established from ...