Human Carotid Artery Endothelial Cells
Cat.No.: CSC-C8594W
Species: Human
Source: Artery
Cell Type: Endothelial Cell
- Specification
- Background
- Scientific Data
- Q & A
- Customer Review
Human carotid artery endothelial cells (HCtAECs) originate from human carotid artery tissue, the major blood vessel between the heart and the head, which brings blood to the brain and head. HCtAECs absorb LDL, are sensitive to platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF).
Among the roles of HCAECs in the functioning of vascular physiology, they regulate endothelial barrier function, proliferate and migrate vascular smooth muscle cells and engage in inflammatory mechanisms. Among AS, HCtAECs is especially pertinent. Atherosclerosis is a multifactorial pathology composed of endothelial dysfunction, inflammatory response, lipid accumulation and others. Studies have found that HCAECs exhibit pro-inflammatory and pro-coagulant characteristics during the formation of atherosclerotic plaques, which are closely related to the stability of the plaques. There are also studies of high levels of homocysteine (Hcy) with pro-inflammatory and pro-coagulant properties during plaque formation and that are tightly linked to plaque stability. Hcy also contributes to the permeability of carotid artery endothelial cells and potentially regulating MCP-1 via the P38/MAPK pathway, which will add to atherosclerosis. To examine how carotid endothelial cells work and become pathogenic, researchers have also developed in vitro culture models, for instance, an injectable carotid artery model made of gelatin scaffolds that mimics a physiological environment is used to study the atherosclerosis process.
NFκB Inhibition Changed Serum Amyloid A-Induced Gene and Inflammatory Protein Expression in Endothelial Cells
The acute phase protein serum amyloid A (SAA), an acute phase protein increased significantly during infection or injury, is implicated in cardiovascular diseases through inducing endothelial dysfunction. It predicts negative outcomes in vascular diseases, promoting inflammation and coagulation. Vallejo et al. explored whether the pharmacological inhibition of NFκB, a transcription factor central to SAA-mediated endothelial inflammation and dysfunction, can protect endothelial function from the detrimental effects induced by SAA.
Human carotid artery endothelial cells (HCtAECs) were treated with the NFκB inhibitor BAY11-7082 before SAA stimulation to assess changes in gene expression and protein activity related to inflammation and coagulation. TF expression is triggered by inflammatory cytokines in atherosclerotic plaques, leading to thrombus formation through coagulation activation at plaque rupture sites. TNFα affects the vascular endothelium, increasing the expression of pro-inflammatory, pro-coagulant, and pro-apoptotic genes. Under normal glucose conditions, HCtAEC exposed to SAA showed significantly higher TNF mRNA levels, but TF mRNA increase was not significant (Fig. 1). Pre-treatment with BAY11-7082 significantly reduced TNF mRNA and moderately inhibited TF mRNA, indicating its effectiveness in countering SAA's pro-inflammatory effects on HCtAEC. Increased TNF expression correlates with IL-6 secretion, indicating cytokine bioactivity. SAA exposure increased IL-6 protein production in HCtAEC (~10-fold). However, BAY11-7082 pre-treatment significantly reduced IL-6 secretion, bringing it below control levels (Fig. 2). Overall, these findings suggest that NFκB inhibition can reduce inflammatory markers.
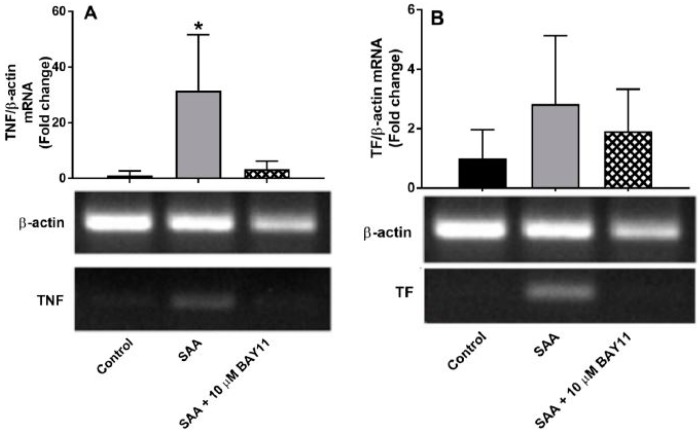 Fig. 1. Relative gene expression in HCtAEC in response to SAA in the absence and presence BAY11-7082 under normoglycaemic conditions (Vallejo A, Chami B, et al., 2018).
Fig. 1. Relative gene expression in HCtAEC in response to SAA in the absence and presence BAY11-7082 under normoglycaemic conditions (Vallejo A, Chami B, et al., 2018).
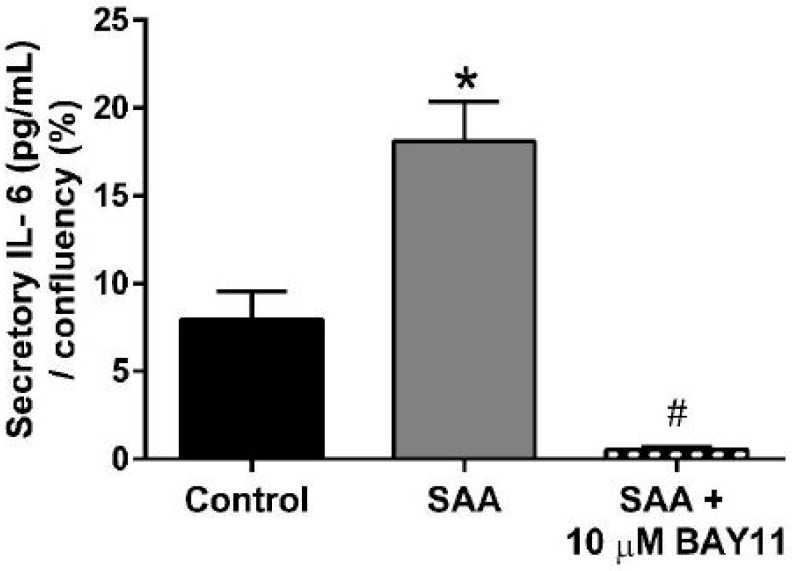 Fig. 2. BAY11-7082 reduces secretory IL-6 induced by SAA. HCtAEC were treated with SAA and BAY11-7082 as defined in methods (Vallejo A, Chami B, et al., 2018).
Fig. 2. BAY11-7082 reduces secretory IL-6 induced by SAA. HCtAEC were treated with SAA and BAY11-7082 as defined in methods (Vallejo A, Chami B, et al., 2018).
Circ_0003204 Knockdown Attenuates Oxidized Low-Density Lipoprotein-Induced Injury via Regulating microRNA-188-3p in Human Carotid Artery Endothelial Cells and THP-1 Cells
Atherosclerosis is a cardiovascular condition caused by inflammation, and in AS, circRNAs and miRNAs regulate cell biology. Circ_0003204, associated with ox-LDL challenge, is suspected to affect AS progression. Oxidised LDL contributes to AS, by activating different cell types such as endothelial and THP-1 cells.
Peng's team used ox-LDL-treated human carotid artery endothelial cells (HCtAECs) and THP-1 cells as models. It tested circ_0003204, miR-188-3p, and TRPC6 expression by dual-luciferase reporter analysis, Western blotting, and other tests to see how the circ_0003204/miR-188-3p/TRPC6 axis affected proliferation, apoptosis, inflammation, and oxidative stress in cells. To determine if circ_0003204 confers ox-LDL-induced HCtAEC damage through miR-188-3p, HCtAECs were transfected with si-con, si-circ_0003204, si-circ_0003204+anti-miR-con, or anti-miR-188-3p prior to ox-LDL exposure. qRT-PCR showed that deletion of circ_0003204 increased miR-188-3p expression in ox-LDL-induced HCtAECs, followed by the inhibition of anti-miR-188-3p (Figure 4A). Circ_0003204 knockdown reversed ox-LDL-induced decreases in cell viability and proliferation that were reduced by miR-188-3p inhibition (Fig. 3B–D). It also suppressed ox-LDL-induced cell death by inhibiting caspase-3 and modulating BAX and BCL2, all blocked by miR-188-3p downregulation (Fig. 3E–I). Additionally, circ_0003204 knockdown in THP-1 cells suppressed ox-LDL-mediated inflammation by decreasing IL-1, IL-6, and TNF-α, followed by miR-188-3p knockdown. It also decreased oxidative stress by lowering MDA and ROS, and elevating SOD, both of which are inhibited by miR-188-3p knockdown (Figures 5D–F). These results indicate that circ_0003204 knockdown reduces ox-LDL-induced damage in HCtAEC and THP-1 cells through miR-188-3p.
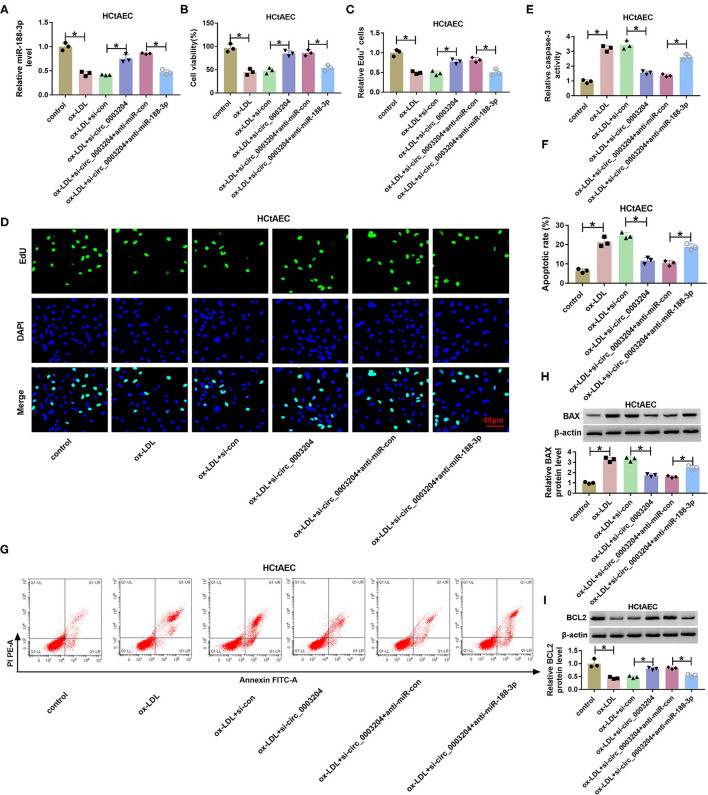 Fig. 3. The effect of circ_0003204 and miR-188-3p on HCtAEC viability and apoptosis (Peng W, Li S, et al., 2021).
Fig. 3. The effect of circ_0003204 and miR-188-3p on HCtAEC viability and apoptosis (Peng W, Li S, et al., 2021).
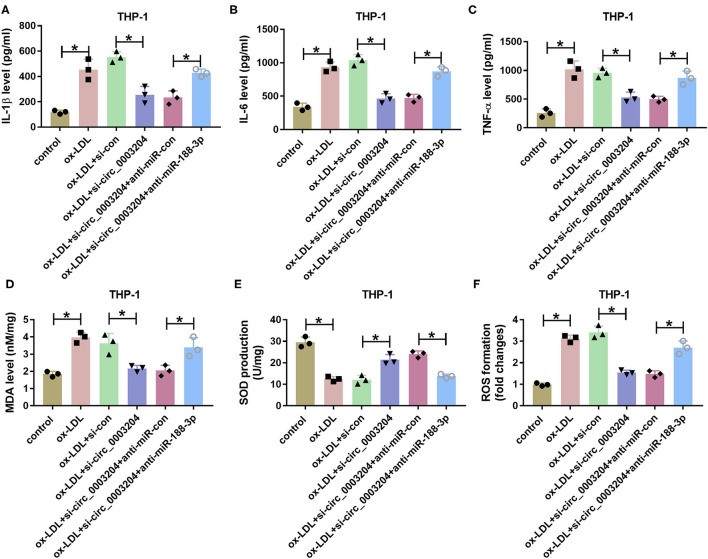 Fig. 4. The effect of circ_0003204 and miR-188-3p on THP-1 cell inflammatory response and oxidative stress (Peng W, Li S, et al., 2021).
Fig. 4. The effect of circ_0003204 and miR-188-3p on THP-1 cell inflammatory response and oxidative stress (Peng W, Li S, et al., 2021).
Ask a Question
Write your own review
Description: Creative Bioarray's Iliac Artery Endothelial Cells, secondary – 500,000 cells per vialSpecial edition cells are isolated from the tissue types described. Cells are sterility and virus tested. All ...

Description: Diseased human pulmonary artery cells and media are convenient and easy to use, allowing the researcher to focus on results. Cryopreserved cells are shipped in third passage. Proliferating cells are ...
Description: Human Primary Coronary Artery Fibroblasts are isolated from normal human coronary artery. Human Primary Coronary Artery Fibroblasts are grown in T75 tissue culture flasks pre-coated with ...

Description: GFP Expressing Human Coronary Artery Endothelial Cells (GFP-HCAECs) provided by Creative Bioarray are puromycin-selected from Human Coronary Artery Endothelial Cells infected with lentivirus ...
Description: Human Internal Thoracic Artery Endothelial Cells are isolated from normal human thoracic artery tissue. The cells are cryopreserved at passage two and delivered frozen. T25 flasks is required for ...
Description: Human hypertension coronary artery fibroblasts are isolated from the coronary artery of human donor that has been diagnosed with hypertension. Human hypertension coronary artery fibroblasts are grown ...