Human Cardiac Microvascular Endothelial Cells
Cat.No.: CSC-C1464
Species: Human
Source: Heart
Cell Type: Endothelial Cell; Microvascular Cell
- Specification
- Background
- Scientific Data
- Q & A
- Customer Review
The vascular endothelium is one of the largest organs in the human body and plays a vital role in hemostasis, gas and nutrient exchange, and regulation of blood flow. In the heart, endothelial cells (ECs) account for a large proportion of non-cardiomyocyte cells, supporting the extensive metabolic demands of the myocardium. Dysfunction of the endothelium in cardiac tissue contributes to the manifestations of heart disease.
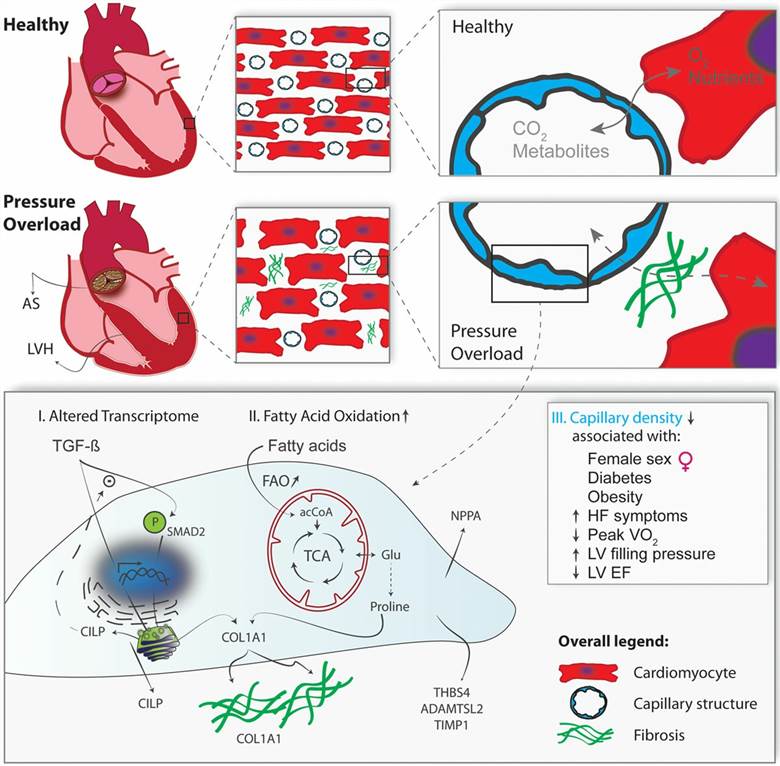 Fig. 1. Pressure overload induces transcriptional, metabolic, and functional changes in cardiac microvascular endothelial cells (Trenson, Sander, et al., 2020).
Fig. 1. Pressure overload induces transcriptional, metabolic, and functional changes in cardiac microvascular endothelial cells (Trenson, Sander, et al., 2020).
Upon chronic pressure overload, interstitial fibrosis, increased stiffness, and hypertrophy of the left ventricle (LV) ultimately lead to heart failure. The failure of recent therapies to curb or revert fibrotic remodeling emphasizes the need for a better understanding of key pathogenic events at different stages of hypertrophic remodeling. Heart failure caused by pressure overload is associated with a distinct molecular phenotypic switch in cardiomyocytes, resulting in increased cardiomyocyte size, protein synthesis, sarcomere reorganization, and re-induction of a fetal gene program. Accumulation of fibroblasts results in excessive extracellular matrix deposition and myocardial stiffness, and the role of TGFβ (transforming growth factor β) in these processes has been extensively studied in cardiomyocytes and fibroblasts. In contrast, genetic fingerprint of microvascular endothelial cells is notoriously lacking, in part because these cells line the smallest branches of the intramyocardial vascular tree and access to these cells is limited. However, microvascular endothelial cells are critically important constituent cells in the heart, located in close proximity to adjacent cardiomyocytes, allowing significant cross talk between both cell types through signal transduction, energy supply, and reciprocal regulation of vasoactive, cytoprotective, growth promoting and proangiogenic molecules. Whereas macrovascular endothelial cell dysfunction is considered the first step in ischemic heart disease, the role of microvascular endothelium in hypertrophic heart disease and the pathogenesis of myocardial fibrosis have not been studied before.
Hypoxia Triggered EndMT in HCMECs
Endothelial-to-mesenchymal transition (EndMT) has been reported in a variety of physiological and pathological phenomena. For example, during heart embryonic development, ECs from endocardium can give rise to the valves and septa of the heart via EndMT. Under the pathological condition, EndMT contributes to many tissue fibrosis diseases such as pulmonary arterial hypertension (PAH), systemic sclerosis (SSc), and unilateral ureteral obstruction (UUO). In particular, EndMT is involved in the development of cardiac fibrosis diseases, such as acute myocardial infarction (AMI), heart failure, and hypertension. It has been shown that the elevation of exogenous and endogenous TGFβ is a potent inducer of EndMT, but accumulating evidence have also suggested that hypoxia, inflammation, and high glucose also induce EndMT.
In order to establish the model of EndMT in vitro, we exposed human cardiac microvascular endothelial cells (HCMECs) to hypoxia condition for 3 days. ECs changed gradually from a cobblestone phenotype to an elongated spindle-shaped morphology (Fig. 1A). RT-PCR analysis (Fig. 1B) showed that hypoxia decreased the expression of CD31 and E-cadherin mRNAs but increased α-SMA, FSP1, and Snail mRNA levels. Compared with the control groups, cells acquired mesenchymal makers (α-SMA, FSP1, and Snail) and lost endothelial makers (CD31 and E-cadherin) (Fig. 1C). Immunofluorescence results (Fig. 1D) further showed that CD31+α-SMA/FSP1+ cells or CD31−α-SMA/FSP1+ cells were found in the hypoxia groups, and CD31+α-SMA/FSP1− cells were found in the normoxia group. All these changes were time dependent (results not shown). Taken together, these data demonstrated that hypoxia induced EndMT.
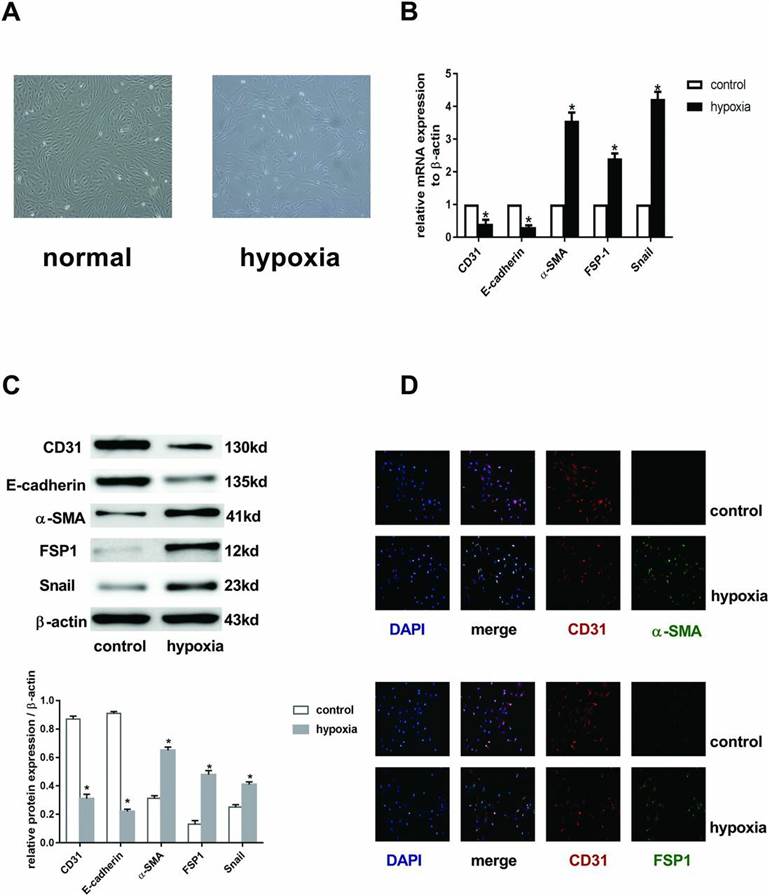 Fig. 1. EndMT was induced by hypoxia in HCMECs (Zou, Jin, et al., 2017).
Fig. 1. EndMT was induced by hypoxia in HCMECs (Zou, Jin, et al., 2017).
Hypoxia Enhances RUNX3 Expression in HCMECs
To investigate the expression of runt-related transcription factor 3 (RUNX3) in HCMECs during hypoxia-induced EndMT, we examined RUNX3 expression by western blotting and RT-qPCR. Both mRNA and protein levels of RUNX3 were low and increased significantly after the cells were cultured in hypoxic conditions for 4 days (Fig. 2 and 3A and B).
To examine whether RUNX3 plays a pivotal role in EndMT of HCMECs, we suppressed RUNX3 expression in HCMECs with RUNX3-RNAi lentivirus and found that RUNX3 knockdown ameliorated hypoxia-induced EndMT (Fig. 4). Suppress of RUNX3 downregulated the mRNA expression of α-SMA and FSP-1 and upregulated the expression of endothelial markers (Fig. 4A). Hypoxia induced a notable increase in the expression of α-SMA and FSP-1, while RUNX3 knockdown attenuated α-SMA and FSP-1 protein expression and increased CD31 and VE-cadherin levels in the HCMECs treated under a hypoxic condition (Fig. 2 and 4B). Double immunofluorescence staining assays were carried out to further confirm that knockdown of RUNX3 attenuated EndMT. CD31 was downregulated while α-SMA was upregulated in the hypoxia group compared with the control group (Fig. 4C), while CD31 was upregulated and α-SMA was downregulated in the RUNX3i group compared with the GFP group (Fig. 4D).
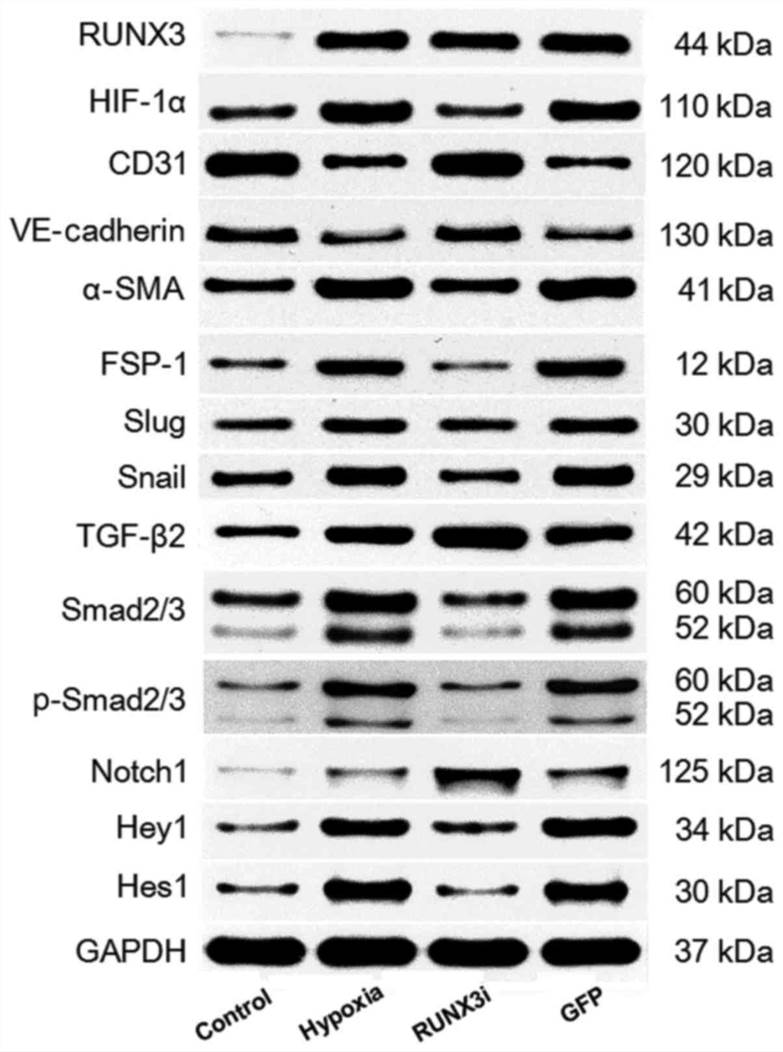 Fig. 2. Western blotting of RUNX3 and its targeting proteins in the control, hypoxia, RUNX3i and green fluorescent protein (GFP) groups (Liu, Yanhua, et al., 2017).
Fig. 2. Western blotting of RUNX3 and its targeting proteins in the control, hypoxia, RUNX3i and green fluorescent protein (GFP) groups (Liu, Yanhua, et al., 2017).
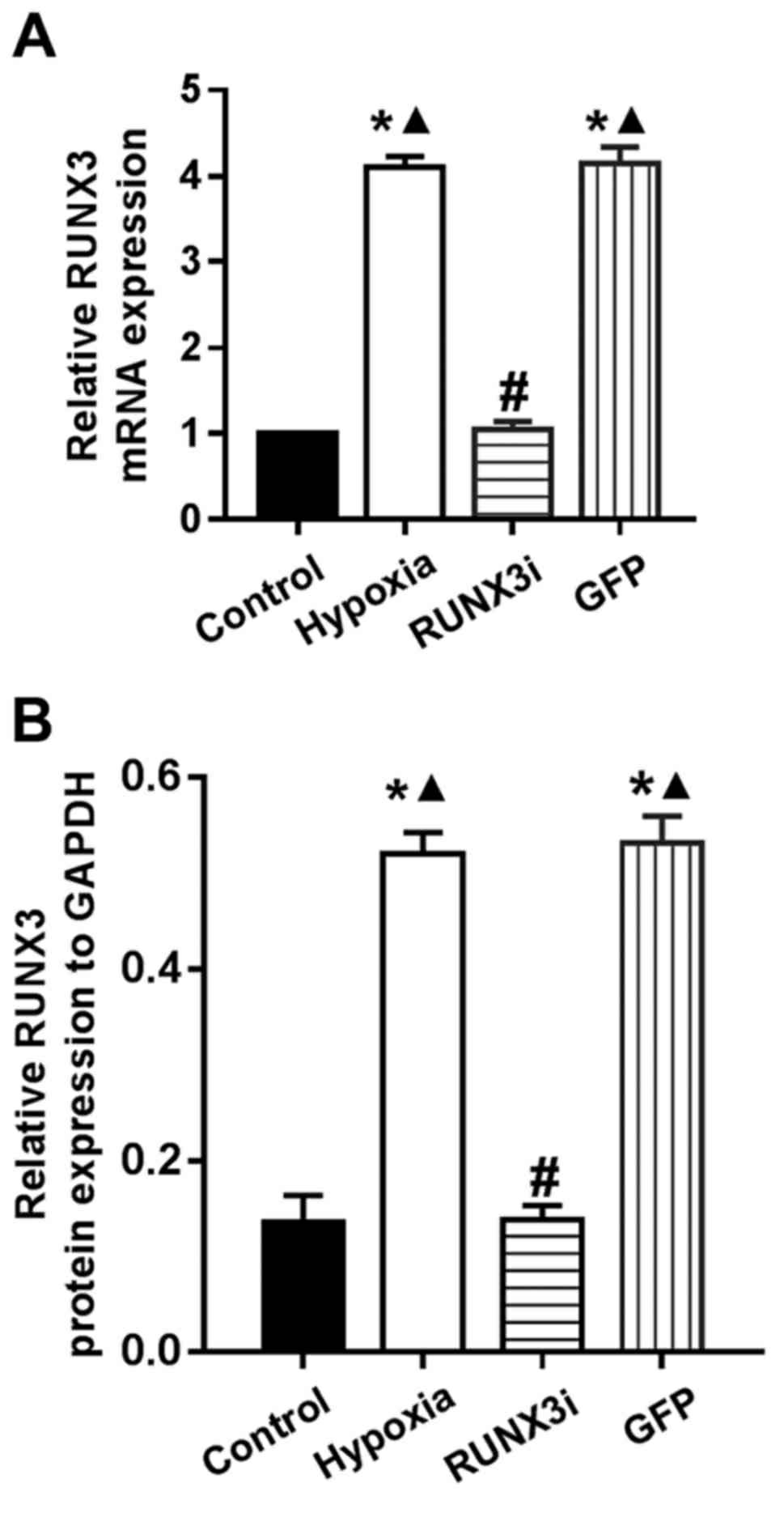 Fig. 3. RT-PCR data showing the mRNA and protein levels of RUNX3 in the control, hypoxia, RUNX3i and green fluorescent protein (GFP) groups (Liu, Yanhua, et al., 2017).
Fig. 3. RT-PCR data showing the mRNA and protein levels of RUNX3 in the control, hypoxia, RUNX3i and green fluorescent protein (GFP) groups (Liu, Yanhua, et al., 2017).
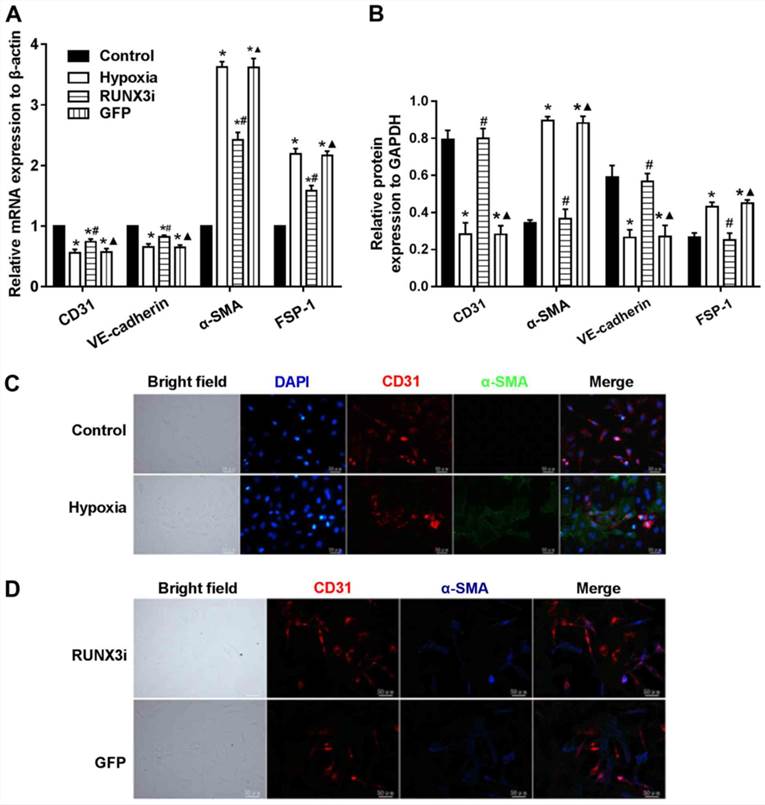 Fig. 4. Knockdown of RUNX3 attenuates EndMT of HCMECs (Liu, Yanhua, et al., 2017).
Fig. 4. Knockdown of RUNX3 attenuates EndMT of HCMECs (Liu, Yanhua, et al., 2017).
Creative Bioarray has a wide variety of microvascular endothelial cell culture media (SuperCult® Microvascular Endothelial Cell Growth Kit-VEGF, SuperCult® Microvascular Endothelial Cell Growth Kit-BBE, or SuperCult® Custom-Made Endothelial Cell Medium) to choose from.
Creative Bioarray's Primary Human Cardiac Microvascular Endothelial Cells are isolated from heart ventricles from a single donor.
Additional donor characteristics including hypertension (cat# CSC-C4985J) are available by contacting our Scientific Support Team.
Check all containers for leakage or breakage. Directly and immediately transfer the cells from dry ice to liquid nitrogen and keep the cells in liquid nitrogen until they are needed for experiments.
Ask a Question
Average Rating: 5.0 | 1 Scientist has reviewed this product
Excellent quality
I recently procured Primary Human Cardiac Microvascular Endothelial Cells from Creative Bioarray, and I must commend the excellent quality of the cells. They are essential for my research and show robust properties and responsiveness.
06 Dec 2023
Ease of use
After sales services
Value for money
Write your own review
Description: HPcF from Creative Bioarray are isolated from the human heart. HPcF are cryopreserved at secondary culture (passage one) and are delivered frozen. Each vial contains >5 x 10^5 cells in 1 ml volume. ...
Description: Human hypertension cardiac microvascular endothelial cells from Creative Bioarray are isolated from the heart of human donor that has been diagnosed with hypertension. Human Hypertension Cardiac ...
Description: HAAF from Creative Bioarray Research Laboratories are isolated from human aortic artery. HAAF are cryopreserved at first passage of primary culture and delivered frozen. Each vial contains >5 x 10^5 ...
Description: Creative Bioarray has developed Human iPSC-Derived Cardiomyocytes, in addition to our human cardiomyocytes isolated from fresh heart tissue. Human iPSC-Derived Cardiomyocytes overcome the limitations ...

Description: HCF from Creative Bioarray Research Laboratories are isolated from the human heart. HCF-a are cryopreserved at secondary culture and delivered frozen. Each vial contains >5 x 10^5 cells in 1 ml ...
Description: Cardiac progenitor cells are derived from the heart (of a single donor) that has been dissociated into single cells and cultured using differential adhesion. These cells are characterized by ...