How to Conduct a Bioavailability Assessment?
Bioavailability describes how much and how quickly a drug or other active material arrives in the circulatory system and has its effects at the point of action after non-intravenous administration (oral administration, inhalation, skin application). That is, absorption ratio (amount) and absorption rate (velocity) is the way to analyze the bioavailability of different formulations. The time it takes a drug to be maximally concentrated in the blood after it has been administered is called Tmax. AUC is the area under the plasma drug concentration-time curve. Bioavailability is most commonly used for the new drugs, changing dosage form or prescription, small therapeutic index drugs.
Bioavailability can be divided into absolute and relative bioavailability. Absolute bioavailability is the percentage of active material infused into the bloodstream when the reference standard is the intravenous dose. This formula is as follows with T and iv being the test solution and reference intravenous solution respectively. D represents the dosage administered.
Fabs=(AUCT·Div) / (AUCiv·DT) × 100%.
Relative bioavailability, on the other hand, compares the bioavailability of a drug across multiple forms in the same circumstances (and where 100% bioavailability has been arbitrarily allocated to a single form). Subscript T and R represent the test preparation and the reference preparation for extravascular administration, respectively, while D represents the administered dose.
Frel=(AUCT·DR) / (AUCR·DT) × 100%
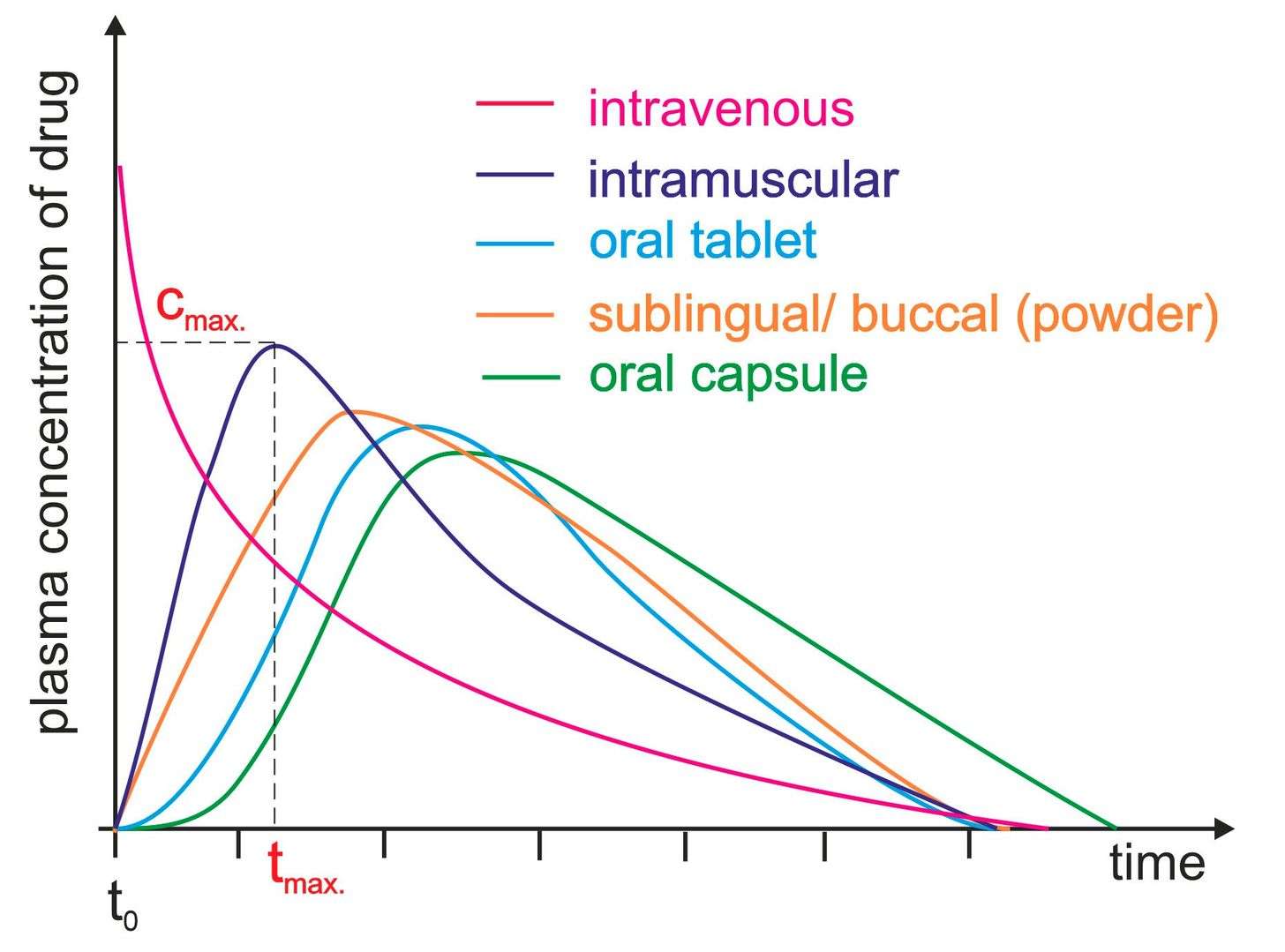 Fig. 1. Plasma level time curves for different types of drug administration. (Stielow, M., Witczyńska, A., et al., 2023).
Fig. 1. Plasma level time curves for different types of drug administration. (Stielow, M., Witczyńska, A., et al., 2023).
Methods of Bioavailability Assessment
Blood concentration method
This approach assesses a drug's pharmacokinetic effects on the body by monitoring the drug concentration in blood over time. It involves several steps, such as blood sampling, plasma concentration determination, and pharmacokinetic calculations. The most important of these is blood sample collection (taking blood samples from subjects within a time frame after the drug has been administered) and the interval for blood collection should be determined by the drug's properties. Blood can be taken from the fingertip, the vein, or the arterial vein. Given the plasma concentration curve, the plasma concentration-time information for drugs can be fitted using the nonlinear least square technique, and some important pharmacokinetic data (pharmacokinetic half-life, plasma maximum, minimum blood concentration, bioavailability of drugs) are available. Such values allow to measure drug absorption, distribution, metabolism and excretion rates, and form a critical starting point for the development and optimization of dosing regimens.
For the following drugs, it is recommended to choose the determination method of blood drug concentration.
- Drugs with narrow therapeutic range and strong toxic side effects.
- Drugs with nonlinear pharmacokinetic properties and dose-dependent elimination rate constant in vivo.
- Drugs requiring long-term use.
- Drugs that need to be used in combination with other medications.
- Drugs where the same dose may result in significant individual differences in blood concentration, leading to substantial pharmacokinetic variability.
Urinary drug data method
Once the drug enters the body, it is excreted from the body primarily via the kidneys in the form of urine. Therefore, the urinary drug data method is an approach to determine the degree of absorption by quantifying the dose of drug excreted in the urine. These steps include administration of drugs, collection of urine at specified times, determination of drug concentration in urine, and calculation of cumulative excretion of drugs. The pharmacokinetic parameters – the urinary excretion rate constant or the total decrementing factor – are then estimated based on the pharmacokinetics, such as the elimination rate constant and the biological half-life.
- Advantages: (1) Urine collection is convenient and minimal harm to the subjects. (2) The drug concentration in urine is higher, allowing for more precise quantitative analysis. (3) The detection method is easier to establish.
- Disadvantages: In the experiment, the average excretion velocity is usually used instead of the instantaneous excretion velocity, which may introduce some errors. Additionally, it requires that most of the drug is excreted in its unchanged form through the urine.
Thus, the urine drug data method is applicable to most drugs excreted in the urine in their original form, especially when the excretion of the drug or its metabolites accounts for more than 70% of the absorbed dose.
Pharmacological effect method
Pharmacological effect analysis evaluates the pharmacokinetic and bioequivalence of a drug by evaluating its pharmacological effects. The standard steps are to calculate the dose-response curve (the pharmacological effect of a drug on the body for different doses) and the time-effect curve (at the same dose, the pharmacological effect of the drug on the body for different times). From these curves we calculate the dose-time curve, which is then used to determine the bioequivalence of drug preparations.
This method is applied when the absorption and speed of drugs are not easy to evaluate by blood drug method and urine drug method, and the effect of drugs is quantitatively related to the amounts of drugs retained in the body, allowing for relatively straightforward quantitative measurement. So, pharmacological effect method is suitable for drugs with complex components and difficult to detect, and can more truly reflect the dynamic change process of drugs in vivo. However, the operation and data analysis of this method are complicated, and the pharmacological effects of some drugs may be affected by multiple factors, which is not easy to measure independently.
Methods for drug concentration measurement
- Spectrophotometry: UV-visible spectrophotometry and fluorescence analysis, etc. This method is simple and sensitive, but has poor specificity. It is suitable for the detection of items with large sample size.
- Chromatography: Techniques such as high-performance liquid chromatography (HPLC), gas chromatography (GC), and mass spectrometry coupled techniques (e.g., LC-MS/MS) fall under this category. These methods are highly sensitive and specific, and is suitable for accurate quantification of trace drugs in complex samples.
- Immunoassay: Including radioimmunoassay (RIA), enzyme immunoassay (EIA) and chemiluminescence immunoassay. These methods are rapid, simple and highly sensitive, especially suitable for the analysis of body fluid samples with low concentration of drugs.
The Main Factors Affecting Bioavailability
- Physicochemical properties of the drug, such as lipid solubility, water solubility, pKa value, molecular size and morphology, stability, etc., which affect the drug's ability to dissolve in the gastrointestinal tract and penetrate biofilms.
- Dosage form factors: Including the characteristics of drug dosage form, such as disintegration time, dissolution rate, and production process, which will affect the release and absorption of drugs in the body.
- Route of Administration: For example, oral drugs must pass through the intestinal wall and enter the portal circulation to reach the liver. During this process, first-pass metabolism may occur, affecting bioavailability.
- Physiological Factors: These involve the effects of gastrointestinal fluids, the transportation of the drug within the gastrointestinal tract, the surface area and local blood flow at the absorption site, the impact of drug metabolism, and intestinal microbiota.
- Liver-mediated metabolic processes, including phase I and phase II metabolism, which may alter drug activity or promote its excretion.
By assessing the bioavailability of a drug, the rate and amount of absorption can be determined to provide a basis for the optimal design of the drug. In addition, bioavailability can also be used to compare the absorption effects of different drug dosage forms and guide the selection and optimization of drug dosage forms.
| Products & Services | Description |
| In Vivo DMPK Services—Bioavailability and Bioequivalence | Creative Bioarray could assist with the study design, protocol development, pharmacokinetic and statistical data assessment, study result report, and raw data preservation. |
Reference
- Stielow, M., et al. The Bioavailability of Drugs-The Current State of Knowledge. Molecules (Basel, Switzerland), 2023. 28(24), 8038.
