Decoding of CYP-Mediated Drug Interactions
Biomolecules. 2024 Jan 12; 14(1): 99.
Authors: Lee J, Beers JL, Geffert RM, Jackson KD.
INTRODUCTION
Drug metabolism is a major determinant of drug concentrations in the body. Drug-drug interactions (DDIs) caused by the co-administration of multiple drugs can lead to alteration in the exposure of the victim drug, raising safety or effectiveness concerns. Assessment of the DDI potential starts with in vitro experiments to determine kinetic parameters and identify risks associated with the use of comedication that can inform future clinical studies. The diverse range of experimental models and techniques has significantly contributed to examining potential DDIs. Cytochrome P450 (CYP) enzymes are responsible for the biotransformation of many drugs on the market, making them frequently implicated in drug metabolism and DDIs. Consequently, there has been a growing focus on the assessment of DDI risk for CYPs.
What Is Reaction Phenotyping?
It is critical to determine if a drug candidate is a substrate for a CYP enzyme before administration to a patient. This is to ensure the drug does not have DDIs that may be of clinical significance. Additionally, knowledge of major metabolizing enzymes can help with any pharmacogenetic, disease state, or environmental considerations. Reaction phenotyping is a commonly used in vitro approach to identify the enzymes and pathways responsible for metabolizing a drug. This is an important step to assess the potential for DDIs as some metabolic pathways may compete for the same enzymes. This type of characterization is recommended for industry sponsors according to the FDA guidance. Understanding the in vitro contributions of CYP enzymes to the overall metabolism of a drug is one of the first steps to determining the need for clinical DDI studies. The main goals of reaction phenotyping are to (1) determine the fraction metabolized by each CYP involved in the metabolic clearance, (2) characterize enzyme kinetics and other in vitro parameters, and (3) provide an early screen for potential DDIs.
Methods for Assessing CYP Inhibition
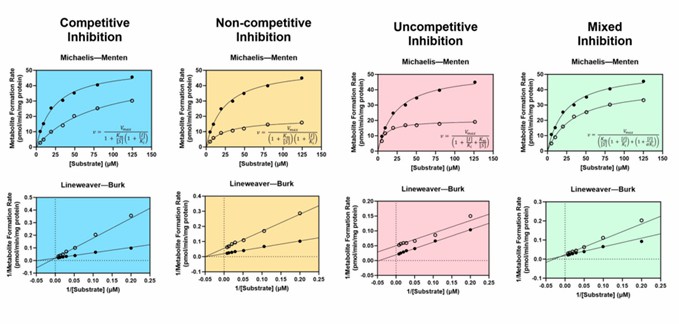 Fig. 1 Enzyme kinetics of reversible inhibition.
Fig. 1 Enzyme kinetics of reversible inhibition.
Assessment of a drug's potential to inhibit CYP enzymes is a multifaceted process that begins early in the preclinical phase of drug development. CYP enzymes typically contain both active and allosteric sites for binding multiple ligands, which may act as substrates, inhibitors, and/or activators. CYP inhibition may be broadly divided into reversible, quasi-irreversible, and irreversible inhibition.
| Early high-throughput screening | Plate-based fluorescent and luminescent assays can be used in early high-throughput screening to determine a drug's inhibitory potential. These experiments typically involve dosing rCYPs in a 96-well format with a pro-fluorescent or pro-luminescent substrate that generates metabolites that can be detected using a plate reader. The IC50 value (the concentration of inhibitor resulting in half-maximal inhibition) may then be calculated by measuring the difference in signal with a range of inhibitor concentrations compared to control incubations. While such assays offer the advantage of high throughput and sensitivity, they are typically only performed with rCYPs since these substrates are not selective for individual CYP enzymes. Furthermore, the assay must reliably yield metabolites with an attached fluorophore to avoid non-specific fluorescence. |
| Probe assays for CYP inhibition | Following initial screening, validated probe assays with HLM are commonly performed before and/or alongside phase I studies. A comprehensive list of both in vitro and clinical index substrates, inhibitors, and inducers for major CYP enzymes and transporters has been published by the U.S. FDA for DDI screening. Importantly, these marker substrate reactions, while useful, are accompanied by several limitations. Although these studies are considered the industry standard for measuring drug inhibition, few (if any) substrates are fully selective for a single CYP enzyme. Often, multiple metabolites are formed simultaneously through multiple pathways, which may affect results if the inhibitor is also metabolized by one or more of the same enzymes as the substrate. |
| Model-based approaches for predicting CYP inhibition | The recent adoption of high throughput screening in DDI assessment has led to the use of predictive modeling to analyze large datasets for potential CYP-mediated DDIs. In silico prediction of CYP-mediated metabolism carries several advantages over traditional in vitro approaches: modeling may be performed very early in drug development to quickly assess many compounds at a low cost. In addition, modeling may be performed with compounds that have yet to be synthesized. |
Methods for Assessing CYP Induction
Unlike CYP inhibition, which can rapidly affect drug metabolism by blocking enzyme activity, CYP enzyme induction is a slower process. CYP induction involves the upregulation of the enzyme biosynthesis, which takes time to reach a new steady-state level. CYP induction by exogenous compounds is mainly mediated by three receptor-dependent mechanisms that can activate transcription of the genes that encode CYP families 1-3. These receptors include aryl hydrocarbon receptor (AhR), which belongs to the basic-helix-loop-helix-Per-Arnt-Sim (bHLH-PAS) family, pregnane X receptor (PXR), and constitutive androstane receptor (CAR). In the absence of ligands, these receptors exist in a latent state in the cytoplasm bound to heat shock protein 90 (Hsp90). When bound to ligands, the receptors undergo a conformational change in the ligand-binding domain, leading to the release of Hsp90, activation, and translocation to the nucleus for transcription.
| Primary human hepatocytes | Cultured primary human hepatocytes express all the relevant hepatic metabolic enzymes, transporters, and their regulators. Among the various in vitro or cell-based approaches, the use of primary hepatocytes is recommended by the FDA and EMA as these cells provide results that are closest to in vivo studies. During drug development, initial experiments can be conducted to evaluate CYP1A2, CYP2B6, and CYP3A4/5. If CYP3A4/5 induction is positive, however, the potential of CYP2C induction by the test drug should be evaluated in the follow-up studies since both CYP3A4/5 and CYP2C enzymes are induced by PXR activation. The incubation duration for inducers typically ranges from 48 to 72 h, which allows the complete induction of enzyme. During the incubation period, the inducer is added daily by replacing the medium containing the drug. The standard endpoints involve the measurement of mRNA levels and/or enzyme activity. |
| Immortalized hepatocytes | Immortalized hepatic cell models offer several advantages for CYP induction studies over primary hepatocytes: easy accessibility, low variability, highly reproducible results in response to inducers, and high availability. Simian virus 40 immortalized human hepatic Fa2N-4 cells express CYP1A2, CYP2C9, and CYP3A4 protein, and the mRNA expression and activity of these enzymes are inducible in response to prototypical inducers. |
| High throughput assays | PXR receptor assays are widely used high throughput assays due to their importance in DDI studies. The cell-free ligand binding assays typically involve quantification of the competition between the test compound and a radiolabeled ligand for receptor binding in genetically expressed and isolated receptor preparations. However, there are instances in which substantial ligand-receptor binding fails to trigger transactivation, resulting in false positives. On the other hand, cell-based transactivation assays are more accurate with fewer false positives, and better correlate with human DDIs. In the transactivation assays, two expression vectors, full-length human PXR and a variation of the target promoter coupled to a reporter such as a luciferase, are transfected into host cells. Host cells are then treated with the test compound, and the reporter gene activity is assayed. Increased reporter gene activity is an indication of induction, and a dose-response curve is generated to determine Emax and EC50. |
Creative Bioarray Relevant Recommendations
| Cat. No. | Product Name |
| DMK-M046 | SuperQuick® Human CYP Inhibition Kit (Mixed) |
| DMK-M047 | SuperQuick® Human CYP Inhibition Kit (CYP1A2) |
| DMK-M048 | SuperQuick® Human CYP Inhibition Kit (CYP2A6) |
| DMK-M049 | SuperQuick® Human CYP Inhibition Kit (CYP3A4) |
| DMK-M050 | SuperQuick® Human CYP Inhibition Kit (CYP2B6) |
View our full range of In Vitro Metabolism Assay Kits and find what you need!
RELATED PRODUCTS & SERVICES

Reference
- Lee J, et al. (2024). "A Review of CYP-Mediated Drug Interactions: Mechanisms and In Vitro Drug-Drug Interaction Assessment." Biomolecules. 14 (1): 99.
