Modeling Human Gastric Cancers in Mice
Cancer Biology & Medicine. 2024 Jun 28; 21 (7): 553-70.
Authors: Zhang W, Wang S, Zhang H, Meng Y, Jiao S, An L, Zhou Z.
INTRODUCTION
Gastric cancer (GC) is a major cause of cancer-related mortality worldwide. GC is determined by multiple (epi)genetic and environmental factors; can occur at distinct anatomic positions of the stomach; and displays high heterogeneity, with different cellular origins and diverse histological and molecular features. This heterogeneity has hindered efforts to fully understand the pathology of GC and develop efficient therapeutics. In the past decade, great progress has been made in the study of GC, particularly in molecular subtyping, investigation of the immune microenvironment, and defining the evolutionary path and dynamics. Preclinical mouse models, particularly immunocompetent models that mimic the cellular and molecular features of human GC, in combination with organoid culture and clinical studies, have provided powerful tools for elucidating the molecular and cellular mechanisms underlying GC pathology and immune evasion, and the development of novel therapeutic strategies.
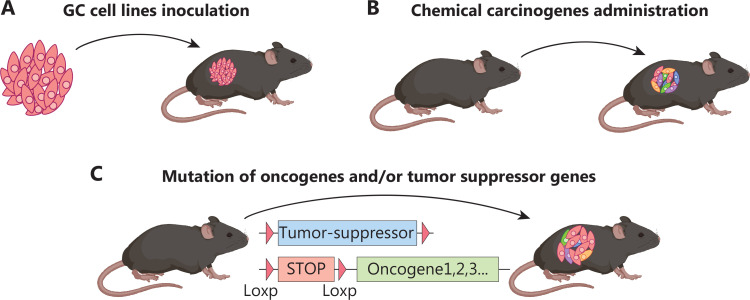 Fig. 1 Immunocompetent mouse models of GC.
Fig. 1 Immunocompetent mouse models of GC.
Non-genetically Engineered GC Mouse Models
Cell line-derived GC graft model
Cell lines derived from patients with GC and mice provide powerful tools to explore the nature of tumor progression and responsiveness to targeted therapy and immunotherapy. In addition to the multiple human GC cell lines available for xenograft study in immunocompromised mice, several mouse GC cell lines (e.g., MFC, MGCC3I, NCC-S1/3, YTN16, and M12), which can be transplanted into immunocompetent mice, have been generated to investigate the molecular and cellular mechanisms governing gastric tumorigenesis and related immune response.
Chemical carcinogen-induced GC mouse models.
MNU, one of the best-characterized chemical carcinogens, can be supplied in the drinking water to induce GC in mice. By introducing alkyl radicals into DNA, MNU causes DNA mutation and dysfunction, thereby promoting gastric tumorigenesis. MNU-induced primary GCs are usually localized in the antrum and involve well to poorly differentiated adenocarcinoma. The tumorigenic efficacy of MNU varies in mice with different genetic backgrounds; male mice on a BALB/c background are relatively susceptible to MNU-induced tumors. In addition, MNU-induced GC is significantly enhanced in combination with other GC risk factors, such as a high-salt diet, H. pylori infection, and Streptococcus anginosus infection. Notably, genetic alterations also significantly influence MNU-dependent tumorigenesis. For example, p53 knockout mice are relatively sensitive to MNU-induced carcinogenesis.
MNNG is another chemical carcinogen particularly widely used in combination with Helicobacter infection to induce GC in mice. MNNG is supplied in the drinking water in 3 cycles at 2-week intervals to induce GC in mice. MNNG-induced primary GC varies across model organisms, including squamous cell carcinoma in the forestomach in mice and adenocarcinomas in the glandular stomach in Mongolian gerbils. Similarly, environmental GC risk factors, including a high-salt diet, a calcium-deficient diet, or catechol, promote the incidence and progression of GC induced by MNNG administration. Moreover, this model has been extensively used to investigate gastric tumorigenesis and targeted therapy against GC.
Genetically Engineered GC Mouse Models
Inflammation-induced GC
- IL-1β transgenic mice. Gastrointestinal cancers are frequently associated with chronic inflammation. For example, chronic inflammation triggered by H. pylori infection or tissue injury in the stomach can initiate sequential histopathologic progression of gastritis to gastric atrophy, intestinal metaplasia, dysplasia, and finally gastric adenocarcinoma. Interleukin-1 polymorphisms have been associated with increased risks of both hypochlorhydrias induced by H. pylori infection and gastric carcinogenesis. H/K-ATPase:hIL-1β transgenic mice expressing secretory human IL-1β specifically in parietal cells have been generated to explore the pathogenic role of hIL-1β during gastric tumorigenesis. These mice spontaneously develop chronic gastritis, hyperplasia, and high-grade dysplasia/adenocarcinoma without invasion into the submucosa or metastasis to distant organs.
- NF-κB1-deficient mice. Deficiency in NF-κB1, even loss of a single allele, can lead to spontaneous intestinal-type gastric adenocarcinoma in mice. Deficiency in NF-κB1 results in increased expression of a variety of inflammatory cytokines, including tumor necrosis factor (TNF), interleukin-6 (IL-6), IL-22, and IL-11, thereby driving aberrant activation of signal transducer and activator of transcription 1 (STAT1). Further genetic depletion of TNF or STAT1 in NF-κB1-deficient mice has been found to prevent invasive GC development.
- Gp130F/F transgenic mice. To investigate the role of dysregulated activation of STAT3 in regulating gastrointestinal epithelial cell homeostasis, it has generated gp130F/F mice by using a phenylalanine knock-in substitution of the IL-6 receptor β-chain Gp130 at the cytoplasmic tyrosine 757 residue, thus preventing its binding to the suppressor of cytokine signaling 3 and enhancing activation of STAT3. Gp130F/F mice spontaneously develop gastric adenoma at the antrum by 4-6 weeks of age, accompanied by splenomegaly and extra-gastric pathologies in the liver and lung. Further knockout of STAT3 in Gp130F/F mice alleviates gastric adenoma progression, thereby highlighting the essential role of STAT3 hyperactivation in GC pathology.
- Transgenic mice with aberrant inflammation induced by T cells. Deregulated T-cell activation mediates gastritis and promotes gastric hyperplasia and adenocarcinomas. For example, T cell-specific deletion of the tumor suppressor liver kinase B1 (LKB1) results in excessive production of proinflammatory cytokines and chemokines such as IL-6, IL-11, and CXCL2, which is accompanied by increased STAT3 activation and infiltration of inflammatory monocytes and neutrophils. The related inflammation promotes the development of gastrointestinal polyposis, a cancer predisposition syndrome. In addition, autoimmune gastritis mediated by self-reactive CD4+ T cells has been found to promote GC development. In a T cell receptor transgenic mouse model of autoimmune gastritis, the T cell receptor targets a peptide from the H+/K+ ATPase proton pump, which is highly expressed on parietal cells in the stomach. Transgenic mice display chronic gastritis with intensive CD4+ T cell infiltration, and elevated IFNγ and IL1-17 production, which is followed by initiation and progression of GC from oxyntic atrophy, mucinous hyperplasia to spasmolytic polypeptide-expressing metaplasia, and intraepithelial neoplasia.
Gastrin/Gastric Acid Disorder-Induced GC
- INS-GAS mice. Gastrin, produced by antrum G cells, is crucial for gastric acid secretion and parietal cell differentiation. A transgenic mouse model termed INS-GAS expressing human gastrin specifically in β islet cells under control of the insulin promoter was originally generated to investigate the potential role of gastrin in regulating islet differentiation. INS-GAS mice show a twofold elevation of serum amidated gastrin and gastrointestinal mucosal hyperplasia. These mice have been further used to examine the role of hypergastrinemia in GC pathology and have shown elevated maximal gastric acid secretion and parietal cell number within 4 months old, but progressive sustained loss of parietal and hypochlorhydria. Eventually, INS-GAS mice develop metaplasia, dysplasia, and invasive GC at 20 months of age.
- Gastrin−/− mice. Gastrin-deficient mice (gastrin−/−) have been generated to investigate the role of gastrin in regulating the development and function of the gastrointestinal tract. Gastrin−/− mice show impaired gastric acid secretion, accompanied by marked abnormalities in gastric gland architecture, with diminished numbers of parietal and enterochromaffin-like cells, and enhanced numbers of mucous neck cells. The loss of parietal cells in gastrin−/− mice has been attributed to bacterial overgrowth and chronic gastritis, and the parietal cell number has been found to normalize after antibiotic treatment. The chronic inflammation resulting from gastric acid secretion disorder in gastrin−/− mice promotes intestinal metaplasia of the gastric epithelium, which eventually develops into polyps by the age of 12 months.
 Fig. 2 Mouse GC models induced by chemical carcinogen administration and/or genetic engineering in immunocompetent mice.
Fig. 2 Mouse GC models induced by chemical carcinogen administration and/or genetic engineering in immunocompetent mice.
GCs Induced by Mutation of Oncogenes And/or Tumor Suppressor Genes
GCs induced by genetic mutations in gastric epithelial cells
- Genetically engineered mice with genetic mutations in pan-epithelial cells. Claudin18.2 is a tight junction membrane protein specifically expressed in the gastric epithelium. Claudin18.2 knockout mice (CLDN18KM) exhibit preneoplastic lesions at 7 weeks and eventually develop high-grade intraepithelial neoplasia at 2 years of age. Mutations in oncogenes and/or tumor suppressor genes specifically in Anxa10+ cells result in mouse GCs with various subtypes possibly mimicking human GC. Knock-in of KrasG12D and Trp53R172H, and deletion of Smad4 in Anxa10+ cells lead to intestinal-type GC, which is prone to metastasis to the liver and the lungs.
- Genetically engineered mice with genetic mutations in stem cells. Lgr5 marks homeostatic stem cells in multiple tissues including the gastrointestinal tract. In the human and mouse stomach, Lgr5 is expressed in a subpopulation of chief cells located at the base of the corpus gland. Lgr5-expressing chief cells drive epithelial renewal after injury and are the cells of origin of GC. Knock-in of KrasG12D or Trp53 deletion in Lgr5-expressing chief cells promotes metaplastic lesions in the corpus. Hyperactivation of the WNT/β-catenin, PI3K, and KRAS signaling pathways by deletion of Apc and Pten and knock-in of KrasG12D in Aqp5+ stem cells cooperatively drive invasive gastric tumorigenesis. Knock-in of KrasG12D and Apc deletion in Mist1-expressing stem cells give rise to intestinal-type metaplasia and cancer.
- Genetically engineered mice with genetic mutations in terminally differentiated cells. Mutations in parietal cells Parietal cells, marked by Atp4b expression, account for one-third of all gastric epithelial cells. Parietal cells secrete gastric acid in response to gastrin stimulation, thereby maintaining the acidic environment of the stomach and inhibiting the invasion of pathogenic microorganisms.
Gastrointestinal stromal tumors (GISTs)
GIST is characterized primarily by activating mutations in KIT or PDGFRA receptor tyrosine kinase. Multiple mouse models of GIST have been established through knock-in of KIT mutations. For example, knock-in of KitV558∆ or KitK641E results in the development of human GIST-like tumors marked by ICC hyperplasia within the myenteric plexus of the GI tract. Multiple mouse models of GIST have been established through knock-in of KIT mutations. For example, knock-in of KitV558∆ or KitK641E results in the development of human GIST-like tumors marked by ICC hyperplasia within the myenteric plexus of the GI tract.
Creative Bioarray Relevant Recommendations
| Service/Product Types | Description |
| Oncology Models | Extensive research has led to the development of powerful animal (most are mice) models of cancer. These models are key tools for current and future cancer research. They allow both the study of normal and abnormal gene interactions in tumors and the reproduction of human disease in mice. |
RELATED PRODUCTS & SERVICES
Reference
- Zhang W, et al. (2024). "Modeling human gastric cancers in immunocompetent mice." Cancer Biol Med. 21 (7): 553-70.
