What factors influence drug distribution?
Pharmacokinetics are the aspects of absorption, distribution, metabolism and excretion that affect how and when a drug reaches a target at the right concentration and time. Clinical pharmacology research has long investigated the connection between drug response and drug concentration, and particularly drug concentration as a measure of pharmacodynamics. The idea is that drugs act primarily by reversible interactions with receptors (typically proteins or enzymes) and disrupt the cellular structure. It is critical to measure the "free" drug concentration at the receptor in a very precise quantitative fashion to establish a correlation between the concentration of drugs and the pharmacological response. The distribution of drugs therefore constitutes the "bridge" between pharmacokinetics and pharmacodynamics and it is the most important factor in pharmacokinetics. But direct quantification of drug flow has been hampered by inaccessibility to target organs and ethical challenges.
Drug distribution is the transport of drugs from the blood supply to body's tissues and organs. The effectiveness and coverage of drug delivery directly influence drug effectiveness and safety. It depends upon a range of variables such as the drug's physicochemical composition, circulation, tissue content and how the drug interacts with macromolecules in the body.
Physicochemical properties of drugs:
The lipid solubility, molecular weight, charge state, and molecular shape of a drug all play key roles in how it distributes throughout the body. Lipid-soluble drugs more easily break through phospholipid layers, and therefore generally stick to lipid-heavy tissues, including the brain, liver and fat tissue. On the other hand, water-soluble drugs tend to be more likely to inhabit aqueous regions, including blood and muscle. It also depends on molecular weight - small molecules can pass faster through biological membranes, and so spread out quickly between multiple organs.
Body Fluid pH:
The concentration of drugs in the body depends on the pH of body fluids. Generally, intracellular fluid pH is around 7.0, and extracellular fluid is about 7.4. Acidic drugs slip better into the alkaline milieu, moving from inside to outside. Conversely, weak basic drugs concentrated in acidic environments can more effectively penetrate cell membranes. Increasing blood pH can facilitate the transfer of weak basic drugs into cells and weak acidic drugs out of cells, such as using sodium bicarbonate to alkalize the blood and urine in phenobarbital poisoning, which aids in moving the drug from brain tissue to plasma and promoting urinary excretion.
Body Barriers:
Certain physiological boundaries, such as the blood-brain barrier and the placental barrier, prevent drug access. The blood-brain barrier between plasma and brain extracellular fluid, or the endothelial-glial barrier between plasma and cerebrospinal fluid (choroid plexus), is the crucial protective layer for brain tissue. By being selectively porous, this barrier prevents the entry and exit of large molecules and water-soluble, particularly dissociable drugs from the central nervous system. But because lipid-soluble medications are able to traverse this barrier, they can more easily access brain tissue.
The placental barrier, made up of placental structures, divides the maternal and foetal blood systems. Highly lipid-soluble medications pass over the placental barrier easily, and highly dissociable medications go over it with greater difficulty. This defence not only shields the foetus from potentially harmful elements, it reduces the therapeutic impact of some key medications.
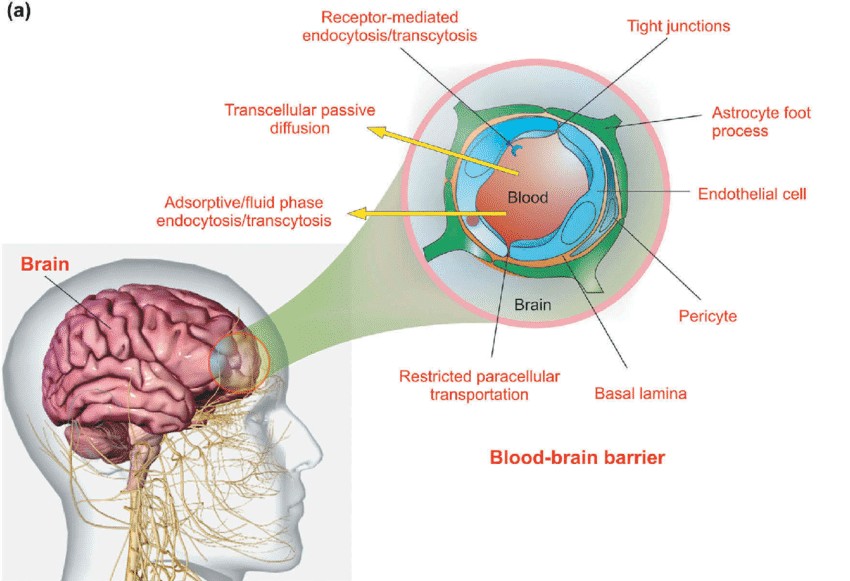 Fig. 1. The schematic illustration of the blood-brain barrier (Jafari B, Pourseif MM, et al., 2019).
Fig. 1. The schematic illustration of the blood-brain barrier (Jafari B, Pourseif MM, et al., 2019).
Tissue Blood Flow and Transporters:
Differences in blood circulation to other tissues greatly impact the rate and extent of drug release. The high-blood-flow organs, including the liver, kidneys and brain, usually peak rapidly. Moreover, higher-flow organs can more readily achieve dynamic equilibrium in the presence of blood-borne substances, while low-flow tissues take longer to reach therapeutic levels. However, in highly perfused tissues such as the liver, spleen, and intestines, although these areas are well-vascularized, drug concentrations may not be high due to the efflux action of transporters. In brain tissue, for example, high levels of P-gp and BCRP transporters significantly limit compound entry into the brain. Similarly, in the liver and kidneys, the functions of these transporters may result in intracellular drug concentrations being higher than plasma free drug concentrations.
Drug-Plasma Protein Binding:
The extent of a drug's binding to plasma proteins determines the proportion present in active form in the bloodstream, typically reducing the concentration of free drug, which may decrease its activity. Drug binding to plasma proteins (such as albumin and α1-acid glycoprotein) is usually reversible, exhibiting temporary inactivity and storage functions. Once saturation of binding sites or competition with other drugs occurs, the concentration of free drug may increase suddenly, leading to enhanced effects or toxic reactions. Studying the kinetics of drug-plasma protein binding can help optimize drug dosage and duration of action.
Drug Interactions with Body Components:
Drugs not only bind to plasma proteins but can also interact with tissue components like proteins in muscle, ligand proteins in liver and kidney cells, or macromolecules such as tubulin, DNA, and mucopolysaccharides within tissues. These interactions affect drug concentration and bioeffects in tissues; drugs bound to tissues, due to difficulty in crossing cell membranes, have higher concentrations in tissues than that of free drug in plasma. For instance, iodine accumulates readily in the thyroid gland, while doxorubicin tends to bind to nuclei with high DNA content. Such selective distribution determines drug potency in different tissues.
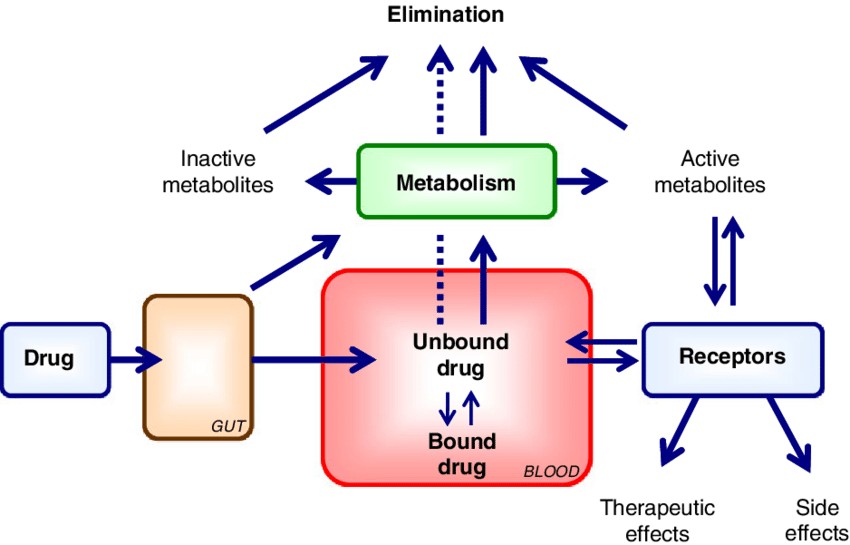 Fig. 2. Protein binding and drug distribution (Vuignier K, Schappler J, et al., 2010).
Fig. 2. Protein binding and drug distribution (Vuignier K, Schappler J, et al., 2010).
Patient Body Weight:
In pharmacokinetic studies, drug distribution within the body is commonly expressed by the parameter volume of distribution (Vd), understood as the hypothetical volume required to achieve a certain drug concentration in plasma or serum. Although the volume of distribution does not correspond directly to a physiologically existing physical space, it provides a practical indicator for assessing the extent of drug distribution within the body. The size of Vd reflects the state of distribution between the blood and body tissues and is closely related to the individual's body weight and metabolic processes.
Knowing how the drugs are distributed is critical to clinical pharmacotherapy. You can control how much drug will get into target tissues by controlling the dose, timing, and delivery route to ensure it gets to target tissues at optimal concentrations with minimum toxicities.
| Products & Services | Description |
| Quantitative Tissue Distribution | Creative Bioarray provides quantitative tissue distribution services to help our customers visualize true tissue distribution, facilitate tissue PK analysis and dosimetry prediction before the initiation of human mass balance studies. |
| In Vivo Blood-Brain-Barrier Assay | Creative Bioarray has established a simple, reliable and efficient in vivo assay that has been successfully applied to several genetic and experimental mouse models. |
| Plasma Protein Binding Assay | Creative Bioarray provides Plasma Protein Binding Assay to help customers accurately detect the binding of compounds to plasma proteins and predict the distribution of compounds in blood and tissues. |
| Blood Plasma Partitioning Assay | Creative Bioarray employs fresh blood samples from humans and various laboratory animals to evaluate a compound's whole blood stability, erythrocyte distribution, and plasma ratio across different species. |
| Brain Tissue Binding Assay | Creative Bioarray provides Brain Tissue Binding Assay to help customers accurately detect the drug transport and distribution in the brain. |
| Microsomal Binding Assay | Creative Bioarray provides Microsomal Binding Assay to help customers accurately understand the relationship between in vitro drug metabolism data and in vivo pharmacokinetics, predicting hepatic clearance and drug-drug interactions. |
References
- Vuignier K, Schappler J, et al. Drug-protein binding: a critical review of analytical tools. Anal Bioanal Chem. 2010. 398(1):53-66.
- Jafari B, Pourseif MM, et al. Peptide-mediated drug delivery across the blood-brain barrier for targeting brain tumors. Expert Opin Drug Deliv. 2019. 16(6):583-605.
